1. DENOMINATION DU MEDICAMENT
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pazopanib (sous forme de chlorhydrate de pazopanib).......................................................... 400 mg
Pour un comprimé pelliculé.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Comprimé pelliculé (comprimé).
Comprimé pelliculé, blanc, en forme de gélule, comportant la mention « 400 » gravée en creux sur une face. Dimensions : 18,0 mm × 7,1 mm environ.
4. DONNEES CLINIQUES
4.1. Indications thérapeutiques
Cancer du rein (Renal Cell Carcinoma RCC)
PAZOPANIB EG est indiqué chez l’adulte en traitement de première ligne du cancer du rein (RCC) avancé et chez les patients préalablement traités par des cytokines à un stade avancé de leur maladie.
Sarcome des tissus mous (Soft Tissue Sarcoma STS)
PAZOPANIB EG est indiqué dans le traitement des patients adultes présentant des sous-types spécifiques de sarcome des tissus mous (STS) avancé, qui ont été préalablement traités par chimiothérapie au stade métastatique ou qui ont progressé dans les 12 mois suivant un traitement (néo) adjuvant.
L’efficacité et la sécurité ont été uniquement établies dans certains sous-types tumoraux histologiques de STS (voir rubrique 5.1).
4.2. Posologie et mode d'administration
Le traitement par PAZOPANIB EG doit être instauré uniquement par un médecin expérimenté dans l’administration de médicaments anticancéreux.
Posologie
Adultes
La dose recommandée de pazopanib dans le traitement du RCC ou du STS est de 800 mg une fois par jour.
Modifications de dose
La dose devra être adaptée (diminuée ou augmentée) par paliers de 200 mg en fonction de la tolérance individuelle au traitement afin de pouvoir gérer les effets indésirables. La dose de pazopanib ne doit pas dépasser 800 mg.
Population pédiatrique
Le pazopanib ne doit pas être utilisé chez les enfants âgés de moins de 2 ans en raison d’un risque potentiel en ce qui concerne la croissance et la maturation des organes (voir rubriques 4.4 et 5.3).
La sécurité et l’efficacité du pazopanib chez les enfants âgés de 2 à 18 ans n’ont pas encore été établies.
Les données actuellement disponibles sont décrites aux rubriques 4.8, 5.1 et 5.2 mais aucune recommandation sur la posologie ne peut être donnée.
Personnes âgées
Les données concernant l’utilisation du pazopanib chez les patients âgés de 65 ans et plus sont limitées. Dans les études portant sur le pazopanib dans le RCC, globalement aucune différence cliniquement significative n’a été observée concernant la sécurité du pazopanib entre les sujets âgés de plus de 65 ans et les sujets plus jeunes. L’expérience clinique n’a pas mis en évidence de différence de réponses entre les personnes âgées et les patients plus jeunes ; cependant, une plus grande sensibilité chez certains patients âgés ne peut être exclue.
Insuffisance rénale
Il est peu probable qu’une insuffisance rénale ait un effet cliniquement pertinent sur la pharmacocinétique du pazopanib compte tenu de la faible excrétion rénale du pazopanib et de ses métabolites (voir rubrique 5.2). Par conséquent, aucune adaptation posologique n’est nécessaire chez les patients ayant une clairance de la créatinine supérieure à 30 mL/min. Il est conseillé d’être prudent chez les patients ayant une clairance de la créatinine inférieure à 30 mL/min étant donné qu’aucune donnée n’est disponible avec le pazopanib dans cette population de patients.
Insuffisance hépatique
Les recommandations posologiques pour les patients présentant une altération de la fonction hépatique ont été établies à partir d’études de pharmacocinétique réalisées avec le pazopanib chez des patients présentant des degrés variables de dysfonctionnement hépatique (voir rubrique 5.2). Des tests de la fonction hépatique doivent être réalisés chez tous les patients, avant l’instauration puis pendant le traitement par pazopanib, afin de déterminer la présence d’une insuffisance hépatique (voir rubrique 4.4). L’administration du pazopanib aux patients présentant une insuffisance hépatique légère ou modérée doit être envisagée avec prudence et impose un suivi attentif de la tolérance au traitement. Huit cents milligrammes de pazopanib une fois par jour constitue la posologie recommandée chez les patients présentant de légères anomalies du bilan hépatique sérique (définies soit par un taux normal de bilirubine associé à une augmentation, quel qu’en soit le degré, du taux d’alanine aminotransférase (ALAT), soit par une augmentation du taux de bilirubine [bilirubine directe > 35 %] jusqu’à 1,5 fois la limite supérieure de la normale (LSN), indépendamment du taux d’ALAT). Une dose réduite de pazopanib à 200 mg une fois par jour est recommandée chez les patients présentant une insuffisance hépatique modérée (définie par une augmentation du taux de bilirubine > 1,5 à 3 × LSN, indépendamment du taux d’ALAT) (voir rubrique 5.2).
Le pazopanib n’est pas recommandé chez les patients présentant une insuffisance hépatique sévère (définie par une bilirubine totale > 3 × LSN, indépendamment du taux d’ALAT).
Voir rubrique 4.4 pour la surveillance hépatique et les modifications posologiques chez les patients présentant une hépatotoxicité d’origine médicamenteuse.
Mode d’administration
PAZOPANIB EG doit être administré par voie orale. Il doit être pris sans nourriture, au moins une heure avant ou deux heures après un repas (voir rubrique 5.2). Les comprimés pelliculés doivent être pris entiers avec de l’eau et non cassés ou écrasés (voir rubrique 5.2).
4.3. Contre-indications
4.4. Mises en garde spéciales et précautions d'emploi
Effets hépatiques
Des cas d’insuffisance hépatique (incluant des décès) ont été rapportés au cours de l’utilisation du pazopanib. L’administration du pazopanib aux patients présentant une insuffisance hépatique légère ou modérée doit être envisagée avec prudence et étroitement surveillée. Huit cents milligrammes de pazopanib une fois par jour constitue la posologie recommandée chez les patients présentant de légères anomalies du bilan hépatique sérique (soit une bilirubine normale et une élévation du taux d’ALAT, quel qu’en soit le degré, ou une augmentation de la bilirubine jusqu’à 1,5 × LSN, indépendamment du taux d’ALAT). Une dose réduite de pazopanib à 200 mg une fois par jour est recommandée chez les patients présentant une insuffisance hépatique modérée (augmentation de la bilirubine > 1,5 à 3 × LSN, indépendamment du taux d’ALAT) (voir rubriques 4.2 et 5.2). Le pazopanib n’est pas recommandé chez les patients présentant une insuffisance hépatique sévère (bilirubine totale > 3 × LSN, indépendamment du taux d’ALAT) (voir rubriques 4.2 et 5.2). Chez ces patients, après administration d’une dose de 200 mg, l’exposition au pazopanib est nettement réduite, bien que cette exposition soit très variable, avec des valeurs considérées comme insuffisantes pour obtenir un effet cliniquement pertinent.
Dans les études cliniques portant sur le pazopanib, des augmentations des transaminases sériques (ALAT, aspartate aminotransférase [ASAT]) et de la bilirubine ont été observées (voir rubrique 4.8). Dans la majorité des cas, des augmentations isolées des ALAT et des ASAT ont été rapportées, sans élévation concomitante des phosphatases alcalines ou de la bilirubine. Les patients âgés de plus de 60 ans peuvent être plus à risque de présenter une augmentation légère (> 3 × LSN) à sévère (> 8 × LSN) des ALAT. Les patients porteurs de l’allèle HLA-B*57:01 ont également un risque accru d’élévations des ALAT associées au pazopanib. La fonction hépatique doit être surveillée chez tous les sujets recevant du pazopanib, quels que soient le génotype ou l’âge (voir rubrique 5.1).
Les tests hépatiques sériques doivent être réalisés avant l’instauration du traitement par pazopanib, aux Semaines 3, 5, 7 et 9, puis aux Mois 3 et 4, avec des examens complémentaires en fonction de l’état clinique du patient. Il conviendra de maintenir un examen périodique après le Mois 4.
Voir le tableau 1 pour les recommandations d’adaptation posologique chez les patients dont les valeurs initiales de la bilirubine totale sont ≤ 1,5 × LSN ; ASAT et ALAT ≤ 2 × LSN :
Tableau 1 Modifications des doses en cas d’hépatotoxicité d’origine médicamenteuse
|
Valeurs des tests de la fonction hépatique |
Modification de dose |
|
Elévation des transaminases comprise entre 3 et 8 × LSN |
Poursuite du pazopanib avec contrôles hebdomadaires de la fonction hépatique jusqu’au retour des transaminases aux valeurs de grade 1 ou aux valeurs initiales. |
|
Elévation des transaminases > 8 × LSN |
Arrêt du pazopanib jusqu’au retour des transaminases aux valeurs de grade 1 ou aux valeurs initiales. Dans le cas où le bénéfice potentiel de la reprise du traitement par pazopanib est estimé supérieur au risque d’hépatotoxicité, le pazopanib peut être réintroduit à une dose réduite de 400 mg par jour et des tests hépatiques sériques doivent être pratiqués sur une base hebdomadaire, pendant 8 semaines. Si, après avoir réintroduit le pazopanib, une élévation des transaminases > 3 × LSN est de nouveau constatée, le traitement par pazopanib doit être définitivement arrêté. |
|
Elévation des transaminases > 3 × LSN avec élévations concomitantes de la bilirubine > 2 × LSN |
Arrêt définitif du pazopanib. Surveillance des patients jusqu’au retour à des valeurs de grade 1 ou aux valeurs initiales. Le pazopanib est un inhibiteur de l’UGT1A1. Une hyperbilirubinémie indirecte (non conjuguée) légère peut survenir chez des patients atteints du syndrome de Gilbert. Les patients ne présentant qu’une hyperbilirubinémie indirecte légère, avec un syndrome de Gilbert connu ou suspecté, et chez lesquels une élévation des ALAT > 3 × LSN est observée doivent être pris en charge conformément aux recommandations établies pour les élévations des ALAT isolées. |
L’utilisation concomitante du pazopanib et de la simvastatine augmente le risque d’élévation des ALAT (voir rubrique 4.5), doit être effectuée avec prudence et faire l’objet d’une surveillance étroite.
Hypertension
Des événements liés à l’hypertension, incluant des épisodes symptomatiques nouvellement diagnostiqués de pression artérielle élevée (crises hypertensives), sont survenus dans les études cliniques portant sur le pazopanib. La pression artérielle devra être bien contrôlée préalablement à l’instauration du traitement par pazopanib. L’hypertension doit être surveillée rapidement après que le traitement a été instauré (au plus tard une semaine après le début du traitement par pazopanib), puis fréquemment par la suite afin de s’assurer du contrôle de la pression artérielle. Des valeurs élevées de pression artérielle (pression artérielle systolique ≥ 150 mmHg ou pression artérielle diastolique ≥ 100 mmHg) ont été rapportées en début de traitement (environ 40 % des cas sont survenus avant le Jour 9 du traitement et environ 90 % des cas sont survenus dans les 18 premières semaines). La pression artérielle doit être surveillée et rapidement prise en charge à la fois par un traitement antihypertenseur et par une modification de la dose de pazopanib (interruption et ré-instauration du traitement à une dose réduite selon l’évaluation clinique) (voir rubriques 4.2 et 4.8). Le traitement par pazopanib doit être arrêté en cas de crise hypertensive avérée ou d’hypertension sévère et persistante malgré un traitement anti-hypertenseur et la réduction des doses de pazopanib.
Syndrome d’encéphalopathie postérieure réversible (PRES)/Syndrome de leucoencéphalopathie postérieure réversible (RPLS)
Des cas de PRES/RPLS ont été rapportés avec le pazopanib. Les PRES/RPLS peuvent se présenter avec des céphalées, une hypertension, des convulsions, une léthargie, une confusion, une cécité et d’autres anomalies visuelles et neurologiques. Ces cas peuvent être d’issue fatale. Le traitement par pazopanib doit être définitivement arrêté chez les patients ayant développé un PRES/RPLS.
Pneumopathie interstitielle diffuse (PID)/pneumopathie inflammatoire
Des cas de PID, parfois d’issue fatale, ont été rapportés avec le pazopanib (voir rubrique 4.8). Les patients devront être surveillés afin de détecter des symptômes pulmonaires pouvant indiquer une PID/ pneumopathie inflammatoire et le traitement par pazopanib devra être arrêté chez les patients développant une PID ou une pneumopathie inflammatoire.
Dysfonctionnement cardiaque/Insuffisance cardiaque
Les risques et bénéfices du pazopanib doivent être pris en considération avant d’instaurer le traitement chez les patients présentant un dysfonctionnement cardiaque préexistant. La sécurité et la pharmacocinétique du pazopanib n’ont pas été étudiées chez les patients présentant une insuffisance cardiaque modérée à sévère et chez ceux dont la valeur de la fraction d’éjection ventriculaire gauche (FEVG) se situe en dessous de la normale.
Des cas de dysfonctionnement cardiaque tels qu’une insuffisance cardiaque congestive et une diminution de la FEVG sont survenus dans les études cliniques réalisées avec le pazopanib (voir rubrique 4.8). Dans une étude randomisée comparant le pazopanib et le sunitinib chez des sujets ayant un RCC (VEG108844), la FEVG a été mesurée à l’inclusion et lors du suivi. Des cas de dysfonctionnement myocardique sont survenus chez 13 % (47/362) des sujets du bras pazopanib et chez 11 % (42/369) des sujets du bras sunitinib. Une insuffisance cardiaque congestive a été observée chez 0,5 % des sujets de chaque bras de traitement. Une insuffisance cardiaque congestive a été rapportée chez 3 des 240 sujets (1 %) dans l’étude STS de phase III VEG110727. Des diminutions de la FEVG ont été détectées chez 11 % (15/140) des sujets contrôlés post-inclusion puis au cours du suivi dans le bras pazopanib versus 3 % (1/39) dans le bras placebo.
Facteurs de risque
Dans l’étude STS de phase III, 13 des 15 patients du bras pazopanib ont présenté une hypertension associée, ce qui a pu aggraver le dysfonctionnement cardiaque chez des patients à risque en augmentant la post-charge cardiaque. 99 % des patients (243/246) inclus dans l’étude STS de phase III, dont les 15 sujets susmentionnés, avaient reçu une anthracycline. Un traitement préalable par anthracycline peut être un facteur de risque de dysfonctionnement cardiaque.
Evolution
Une évolution favorable (plus ou moins 5 % des valeurs initiales) a été rapportée chez 4 des 15 sujets et une normalisation partielle (dans l’intervalle des valeurs normales mais > 5 % en dessous de leurs valeurs initiales) chez 5 sujets. L’évolution n’a pas été favorable chez 1 sujet et des données de suivi n’étaient pas disponibles pour les 5 autres sujets.
Prise en charge
Une interruption du traitement par pazopanib et/ou une diminution de la dose de pazopanib doivent être associées au traitement de l’hypertension (en cas d’hypertension, se référer aux mises en garde ci-dessus) chez les patients dont la FEVG est significativement diminuée, si le tableau clinique le justifie.
Les patients doivent être étroitement surveillés afin de détecter d’éventuels signes ou symptômes cliniques d’une insuffisance cardiaque congestive. Un contrôle de la FEVG à l’instauration du traitement, puis régulièrement, est recommandé chez les patients à risque de dysfonctionnement cardiaque.
Allongement de l’intervalle QT et torsades de pointes
Dans les études cliniques portant sur le pazopanib, des événements correspondant à un allongement de l’intervalle QT et des torsades de pointes sont survenus (voir rubrique 4.8). Le pazopanib doit être utilisé avec prudence chez les patients présentant des antécédents d’allongement de l’intervalle QT, chez les patients prenant des antiarythmiques ou d’autres médicaments pouvant allonger l’intervalle QT et chez les patients présentant une maladie cardiaque préexistante significative. Sous pazopanib, la surveillance par des électrocardiogrammes à l’inclusion et régulièrement, et le maintien des électrolytes (par exemple, calcium, magnésium, potassium) dans les valeurs normales sont recommandés.
Evénements thrombotiques artériels
Dans les études cliniques portant sur le pazopanib, infarctus du myocarde, ischémie myocardique, accident ischémique cérébral et accident ischémique transitoire ont été observés (voir rubrique 4.8). Des événements d’issue fatale ont été observés. Le pazopanib doit être utilisé avec prudence chez les patients présentant un risque accru d’événements thrombotiques ou ayant eu des antécédents d’événements thrombotiques. Le pazopanib n’a pas été étudié chez les patients ayant eu un événement dans les 6 mois précédant son instauration. La décision de traiter doit être prise sur la base de l’évaluation du rapport bénéfice/risque de chaque patient.
Evénements thromboemboliques veineux
Dans les études cliniques portant sur le pazopanib, des événements thromboemboliques veineux tels que thrombose veineuse et embolie pulmonaire d’issue fatale sont survenus. Bien qu’observés dans les études RCC et STS, leur incidence était plus élevée dans la population STS (5 %) que dans la population RCC (2 %).
Microangiopathie thrombotique (MAT)
Des cas de MAT ont été rapportés dans les études cliniques portant sur le pazopanib en monothérapie, en association avec le bévacizumab, ainsi qu’en association avec le topotécan (voir rubrique 4.8). Les patients présentant une MAT doivent définitivement arrêter le traitement par pazopanib. Une réversibilité des effets de MAT a été observée après arrêt du traitement. Le pazopanib n’est pas indiqué en association avec d’autres agents.
Evénements hémorragiques
Dans les études cliniques portant sur le pazopanib, des événements hémorragiques ont été rapportés (voir rubrique 4.8). Des événements hémorragiques d’issue fatale sont survenus. Le pazopanib n’a pas été étudié chez les patients présentant des antécédents d’hémoptysie, d’hémorragie cérébrale, ou d’hémorragie gastro-intestinale (GI) cliniquement significative survenues au cours des 6 derniers mois. Le pazopanib doit être utilisé avec prudence chez les patients présentant des risques significatifs d’hémorragie.
Anévrismes et dissections artérielles
L’utilisation d’inhibiteurs des voies du VEGF chez les patients souffrant ou non d’hypertension peut favoriser la formation d’anévrismes et/ou de dissections artérielles. Avant l’instauration du pazopanib, ce risque doit être soigneusement pris en considération chez les patients présentant des facteurs de risque tels que l’hypertension ou des antécédents d’anévrisme.
Perforations et fistules gastro-intestinales (GI)
Dans les études cliniques portant sur le pazopanib, des événements correspondant à une perforation ou une fistule GI sont survenus (voir rubrique 4.8). Des événements de perforations d’issue fatale se sont produits. Le pazopanib doit être utilisé avec prudence chez les patients à risque de perforation ou de fistule GI.
Cicatrisation des plaies
Aucune étude spécifique concernant l’effet du pazopanib sur la cicatrisation des plaies n’a été menée. Etant donné que les inhibiteurs du facteur de croissance endothélial vasculaire (VEGF) peuvent altérer la cicatrisation des plaies, le traitement par pazopanib devra être arrêté au moins 7 jours avant une intervention chirurgicale programmée. La décision de reprendre le traitement par pazopanib après une intervention chirurgicale doit reposer sur le constat clinique d’une cicatrisation appropriée des plaies. Le traitement par pazopanib doit être arrêté chez les patients présentant une déhiscence de la plaie.
Hypothyroïdie
Dans les études cliniques portant sur le pazopanib, des événements d’hypothyroïdie sont survenus (voir rubrique 4.8). Avant le début du traitement par pazopanib, des dosages biologiques de la fonction thyroïdienne à l’inclusion sont recommandés et les patients atteints d’hypothyroïdie doivent être traités conformément à la pratique médicale standard. Les signes et symptômes d’un dysfonctionnement thyroïdien doivent être étroitement surveillés chez tous les patients traités par pazopanib.
Une surveillance biologique de la fonction thyroïdienne doit être réalisée régulièrement et toute anomalie doit être prise en charge conformément à la pratique médicale standard.
Protéinurie
Dans les études cliniques portant sur le pazopanib, des cas de protéinurie ont été rapportés. Il est recommandé de réaliser une analyse urinaire à l’inclusion et régulièrement pendant le traitement, et de surveiller l’aggravation d’une protéinurie. Le traitement par pazopanib doit être arrêté si le patient développe un syndrome néphrotique.
Syndrome de lyse tumorale (SLT)
La survenue d’un SLT, y compris un SLT d’issue fatale, a été associée à l’utilisation du pazopanib (voir rubrique 4.8). Les patients présentant un risque plus élevé de SLT sont ceux présentant une évolution tumorale rapide, une charge tumorale élevée, un dysfonctionnement rénal ou une déshydratation. Des mesures préventives, telles que les traitements par des taux élevés d’acide urique et l’hydratation par voie intraveineuse, doivent être envisagées avant l’instauration du pazopanib. Les patients à risque doivent être surveillés étroitement et pris en charge conformément à leur tableau clinique.
Pneumothorax
Dans les études cliniques portant sur le pazopanib menées chez des patients présentant un sarcome des tissus mous avancé, des événements de pneumothorax sont survenus (voir rubrique 4.8). Les patients traités par pazopanib doivent être étroitement surveillés afin de détecter d’éventuels signes et symptômes d’un pneumothorax.
Population pédiatrique
Le mécanisme d’action du pazopanib pouvant sévèrement affecter la croissance et la maturation des organes au cours du développement postnatal précoce chez le rongeur (voir rubrique 5.3), le pazopanib ne doit pas être administré aux patients pédiatriques âgés de moins de 2 ans.
Infections
Des cas d’infections graves (avec ou sans neutropénie), parfois d’issue fatale, ont été rapportés.
Association à d’autres traitements anticancéreux systémiques
Des études cliniques portant sur le pazopanib en association avec un certain nombre d'autres traitements anticancéreux (y compris, par exemple, le pémétrexed, le lapatinib ou le pembrolizumab) ont été prématurément arrêtées en raison d’une augmentation de la toxicité et/ou de la mortalité. Une dose bien tolérée et efficace n’a pas été établie avec ces schémas.
Grossesse
Les études pré-cliniques chez l’animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3). Si le pazopanib est utilisé pendant la grossesse, ou si la patiente débute une grossesse au cours du traitement par pazopanib, elle devra être informée des risques potentiels pour le fœtus. Il est conseillé aux femmes en âge de procréer d’éviter de débuter une grossesse pendant le traitement par pazopanib (voir rubrique 4.6).
Interactions
L’administration concomitante avec des inhibiteurs puissants du CYP3A4, de la glycoprotéine P (P-gp) ou de la protéine de résistance au cancer du sein (BCRP) doit être évitée en raison du risque d’augmentation de l’exposition au pazopanib (voir rubrique 4.5). L’utilisation d’un autre traitement pris de façon concomitante et présentant un potentiel inhibiteur minimal ou nul du CYP3A4, de la P-gp ou de la BCRP doit être envisagée.
L’administration concomitante avec des inducteurs du CYP3A4 doit être évitée en raison du risque de diminution de l’exposition au pazopanib (voir rubrique 4.5).
Des cas d’hyperglycémie ont été observés au cours d’un traitement concomitant par kétoconazole.
L’administration concomitante de pazopanib avec des substrats de l’uridine diphosphate glucuronosyl transférase 1A1 (UGT1A1) (par exemple, irinotécan) doit être envisagée avec prudence étant donné que le pazopanib est un inhibiteur de l’UGT1A1 (voir rubrique 4.5).
Le jus de pamplemousse devra être évité pendant le traitement par pazopanib (voir rubrique 4.5).
Excipients
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé pelliculé, c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Effets des autres médicaments sur le pazopanib
Les études in vitro suggèrent que le métabolisme oxydatif du pazopanib dans les microsomes hépatiques humains est médié principalement par le CYP3A4, avec des contributions mineures des CYP1A2 et CYP2C8. Par conséquent, les inhibiteurs et les inducteurs du CYP3A4 peuvent altérer le métabolisme du pazopanib.
Inhibiteurs du CYP3A4, de la P-gp et de la BCRP
Le pazopanib est un substrat pour le CYP3A4, la P-gp et la BCRP.
L’administration concomitante du pazopanib (400 mg une fois par jour) avec le kétoconazole (400 mg une fois par jour), un inhibiteur puissant du CYP3A4 et de la P-gp, pendant 5 jours consécutifs, a entraîné une augmentation de l’ASC(0-24) et de la Cmax moyennes du pazopanib de respectivement 66 % et 45 %, par rapport à l’administration du pazopanib seul (400 mg une fois par jour pendant 7 jours). La comparaison des paramètres pharmacocinétiques de la Cmax (moyennes comprises entre 27,5 et 58,1 μg/mL) et de l’ASC(0-24) (moyennes comprises entre 48,7 et 1 040 μg*h/mL) du pazopanib après administration de 800 mg de pazopanib seul et après administration de 400 mg de pazopanib plus 400 mg de kétoconazole (Cmax moyenne de 59,2 μg/mL, ASC(0-24) moyenne de 1 300 μg*h/mL) a indiqué que, en présence d’un inhibiteur puissant du CYP3A4 et de la P-gp, une réduction de la dose de pazopanib à 400 mg une fois par jour entraînera, chez la majorité des patients, une exposition systémique similaire à celle observée après administration de 800 mg de pazopanib seul, une fois par jour. Toutefois, l’exposition systémique au pazopanib peut, chez certains patients, s’avérer plus importante que celle observée après administration de 800 mg de pazopanib seul.
L’administration concomitante du pazopanib avec d’autres inhibiteurs puissants de la famille du CYP3A4 (par exemple, itraconazole, clarithromycine, atazanavir, indinavir, néfazodone, nelfinavir, ritonavir, saquinavir, télithromycine, voriconazole) peut augmenter les concentrations de pazopanib. Le jus de pamplemousse contient un inhibiteur du CYP3A4 et peut également augmenter les concentrations plasmatiques de pazopanib.
L’administration de 1 500 mg de lapatinib (substrat et faible inhibiteur du CYP3A4 et de la P-gp et puissant inhibiteur de la BCRP) avec 800 mg de pazopanib a entraîné une augmentation approximative de 50 % à 60 % de l’ASC(0-24) et de la Cmax moyenne du pazopanib par rapport à l’administration de 800 mg de pazopanib seul.
L’inhibition de la P-gp et/ou de la BCRP par le lapatinib a probablement contribué à l’augmentation de l’exposition au pazopanib.
L’administration concomitante du pazopanib et d’un inhibiteur du CYP3A4, de la P-gp et de la BCRP, tel que le lapatinib, entraînera une augmentation des concentrations plasmatiques de pazopanib. L’administration concomitante avec des inhibiteurs puissants de la P-gp ou de la BCRP peut également altérer l’exposition et la distribution du pazopanib, incluant la distribution dans le système nerveux central (SNC).
L’utilisation concomitante du pazopanib avec un inhibiteur puissant du CYP3A4 doit être évitée (voir rubrique 4.4). Dans le cas où l’association avec un inhibiteur puissant du CYP3A4 ne pourrait être remplacée par aucune alternative médicalement acceptable, la dose du pazopanib devra être réduite à 400 mg une fois par jour tout au long de l’administration concomitante. Dans de tels cas, les effets indésirables doivent être étroitement surveillés et des diminutions supplémentaires de la posologie peuvent être envisagées si d’éventuels événements indésirables liés au médicament sont observés.
L’association aux inhibiteurs puissants de la P-gp ou de la BCRP doit être évitée, ou l’utilisation d’un autre médicament pris de façon concomitante et présentant un potentiel inhibiteur minimal ou nul de la P-gp ou de la BCRP est recommandée.
Inducteurs du CYP3A4, de la P-gp et de la BCRP
Les inducteurs du CYP3A4 comme la rifampicine peuvent diminuer les concentrations plasmatiques de pazopanib. L’administration concomitante du pazopanib avec des inducteurs puissants de la P-gp ou de la BCRP peuvent altérer l’exposition et la distribution du pazopanib, incluant la distribution dans le SNC. L’utilisation d’un autre traitement pris de façon concomitante et ne présentant pas ou peu d’effet inducteur d’enzyme ou de transporteur est recommandée.
Effets du pazopanib sur d’autres médicaments
Les études in vitro avec des microsomes hépatiques humains ont révélé que le pazopanib inhibait les enzymes CYP 1A2, 3A4, 2B6, 2C8, 2C9, 2C19 et 2E1. L’induction potentielle du CYP3A4 humain a été démontrée lors d’études in vitro du récepteur PXR humain. Les études de pharmacologie clinique, utilisant 800 mg de pazopanib une fois par jour, ont démontré que le pazopanib n’avait pas d’effet cliniquement significatif sur la pharmacocinétique de la caféine (substrat de test du CYP1A2), de la warfarine (substrat de test du CYP2C9), ou de l’oméprazole (substrat de test du CYP2C19) chez les patients atteints de cancer. L’administration de pazopanib a entraîné une augmentation de 30 % environ de l’ASC et de la Cmax moyennes du midazolam (substrat de test du CYP3A4) et une augmentation de 33 % à 64 % du rapport des concentrations urinaires du dextrométhorphane/dextrophane après administration par voie orale de dextrométhorphane (substrat de test du CYP2D6). L’administration concomitante de 800 mg de pazopanib une fois par jour et de 80 mg/m2 de paclitaxel (substrat du CYP3A4 et du CYP2C8) une fois par semaine a entraîné une augmentation moyenne de l’ASC et de la Cmax du paclitaxel, respectivement de 26 % et 31 %.
Sur la base des valeurs de l’IC50 in vitro et de la Cmax plasmatique in vivo, il a été mis en évidence que les métabolites GSK1268992 et GSK1268997 du pazopanib pouvaient contribuer à l’effet inhibiteur net du pazopanib vis-à-vis de la BCRP. De plus, l’inhibition de la BCRP et de la P-gp par le pazopanib dans le tractus gastro-intestinal ne peut être exclue. Une attention particulière doit être portée lors de l’administration du pazopanib de façon concomitante avec des substrats oraux de la BCRP et de la P-gp.
In vitro, le pazopanib a inhibé le polypeptide de transport d’anion organique humain (OATP1B1). Il ne peut être exclu que le pazopanib affectera la pharmacocinétique des substrats de l’OATP1B1 (par exemple, statines, voir « Effet de l’utilisation concomitante du pazopanib et de la simvastatine » ci-dessous).
In vitro, le pazopanib est un inhibiteur de l’enzyme uridine diphosphoglucuronosyl-transférase 1A1 (UGT1A1). Le métabolite actif de l’irinotécan, SN-38, est un substrat de l’OATP1B1 et de l’UGT1A1. La co-administration de 400 mg de pazopanib une fois par jour avec le cétuximab (250 mg/m2) et l’irinotécan (150 mg/m2) a entraîné une augmentation de l’exposition systémique au SN-38 d’environ 20 %. L’impact du pazopanib sur la biodisponibilité du SN-38 peut être majoré chez les patients présentant le polymorphisme UGT1A1*28 comparé aux sujets porteurs de l’allèle de type sauvage. Toutefois, le génotype UGT1A1 n’est pas systématiquement prédictif de la biodisponibilité du SN-38. L’administration du pazopanib en association avec les substrats de l’UGT1A1 doit faire l’objet d’une attention particulière.
Effet de l’utilisation concomitante du pazopanib et de la simvastatine
L’utilisation concomitante du pazopanib et de la simvastatine augmente l’incidence des élévations de l’ALAT. Les résultats d’une méta-analyse utilisant des données combinées d’études cliniques réalisées avec le pazopanib révèlent que des taux d’ALAT > 3 × LSN ont été rapportés chez 126/895 (14 %) des patients n’utilisant pas de statines, par rapport à 11/41 (27 %) des patients co-traités par simvastatine (p = 0,038). Si un patient recevant un traitement concomitant par simvastatine développe des élévations du taux d’ALAT, se référer aux recommandations relatives à la posologie du pazopanib et interrompre le traitement par simvastatine (voir rubrique 4.4). De plus, l’utilisation concomitante du pazopanib et d’autres statines doit être effectuée avec prudence, les données disponibles n’étant pas suffisantes pour évaluer leur impact sur les taux d’ALAT. Il ne peut être exclu que le pazopanib modifie la pharmacocinétique d’autres statines (par exemple, atorvastatine, fluvastatine, pravastatine, rosuvastatine).
Effet de la nourriture sur le pazopanib
L’administration de pazopanib avec un repas riche ou pauvre en matières grasses a entraîné une augmentation de l’ASC et de la Cmax d’un facteur 2 environ. Par conséquent, le pazopanib doit être administré au moins 1 heure avant ou 2 heures après un repas.
Médicaments augmentant le pH gastrique
L’administration concomitante de pazopanib et d’ésoméprazole diminue la biodisponibilité du pazopanib d’environ 40 % (ASC et Cmax) et la co-administration de pazopanib avec des médicaments qui augmentent le pH gastrique doit être évitée. Dans le cas où l’utilisation concomitante d’un inhibiteur de la pompe à protons (IPP) s’avère médicalement nécessaire, il est recommandé que la dose de pazopanib soit prise une fois par jour le soir, sans nourriture, au même moment que l’IPP. Dans le cas où l’administration concomitante d’un antagoniste du récepteur H2 s’avère médicalement nécessaire, le pazopanib doit être pris sans nourriture, au moins 2 heures avant ou au moins 10 heures après l’administration de l’antagoniste du récepteur H2.
Le pazopanib doit être administré au moins 1 heure avant ou 2 heures après l’administration d’antiacides d’action rapide. Les recommandations sur les modalités de co-administration des IPP et des antagonistes du récepteur H2 reposent sur des considérations physiologiques.
4.6. Fertilité, grossesse et allaitement
Grossesse/Contraception chez les hommes et les femmes
Il n’existe pas de données pertinentes concernant l’utilisation du pazopanib chez la femme enceinte. Les études effectuées chez l’animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3). Le risque potentiel chez l’être humain n’est pas connu.
Le pazopanib ne doit pas être utilisé pendant la grossesse à moins que l’état clinique de la femme ne nécessite un traitement par pazopanib. Si le pazopanib est utilisé pendant la grossesse, ou si la patiente débute une grossesse au cours du traitement par pazopanib, elle devra être informée des risques potentiels pour le fœtus.
Il est recommandé aux femmes en âge de procréer d’avoir recours à une méthode efficace de contraception pendant le traitement et pendant au moins 2 semaines après la dernière dose de pazopanib et d’éviter de débuter une grossesse pendant le traitement par pazopanib.
Les patients masculins (y compris ceux ayant eu une vasectomie) doivent utiliser des préservatifs lors des rapports sexuels pendant le traitement par pazopanib et pendant au moins 2 semaines après la dernière dose de pazopanib pour éviter toute exposition potentielle au médicament à leurs partenaires enceintes ou leurs partenaires féminines en âge de procréer.
Allaitement
L’innocuité du pazopanib pendant l’allaitement n’a pas été établie. Aucune donnée sur le passage du pazopanib ou de ses métabolites dans le lait maternel n’est disponible. Il n’existe pas de données chez l’animal sur l’excrétion du pazopanib dans le lait animal. Un risque pour l’enfant allaité ne peut être exclu. L’allaitement doit être interrompu au cours du traitement par pazopanib.
Fertilité
Les études effectuées chez l’animal indiquent que la fertilité mâle et femelle peut être affectée par un traitement par pazopanib (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
4.8. Effets indésirables
Résumé du profil de sécurité
L’évaluation globale de la sécurité et de la tolérance du pazopanib (total : n = 1 149) chez les patients ayant un RCC se base sur les données combinées de l’étude pivot (VEG105192, n = 290), de l’étude d’extension (VEG107769, n = 71), de l’étude de soutien de phase II (VEG102616, n = 225) et de l’étude de non infériorité de phase III, randomisée, en ouvert et en groupes parallèles (VEG108844, n = 557) (voir rubrique 5.1).
Les données combinées de l’étude STS pivot (VEG110727, n = 369) et de l’étude de soutien de phase II (VEG20002, n = 142) ont été analysées dans le cadre de l’évaluation globale de sécurité et de tolérance du pazopanib (total de la population de sécurité n = 382) chez les sujets ayant un STS (voir rubrique 5.1).
Les effets indésirables graves les plus importants identifiés dans les études RCC ou STS et rapportés chez < 1 % des patients traités étaient accident ischémique transitoire, accident ischémique cérébral, ischémie myocardique, infarctus du myocarde et infarctus cérébral, dysfonctionnement cardiaque, perforation et fistule gastro-intestinales, allongement de l’intervalle QT, torsades de pointes et hémorragies pulmonaires, gastro-intestinales et cérébrales. D’autres effets indésirables graves importants identifiés dans les études STS ont inclus des événements thromboemboliques veineux, un dysfonctionnement ventriculaire gauche et un pneumothorax.
Les événements d’issue fatale, considérés comme possiblement liés au pazopanib, incluaient : hémorragie gastro-intestinale, hémorragie pulmonaire/hémoptysie, fonction hépatique anormale, perforation intestinale et accident ischémique cérébral.
Dans les essais RCC et STS, les effets indésirables les plus fréquents (survenus chez au moins 10 % des patients) et de tout grade ont notamment été : diarrhée, modification de la couleur des cheveux, hypopigmentation cutanée, rash exfoliatif, hypertension, nausées, céphalées, fatigue, anorexie, vomissements, dysgueusie, stomatite, poids diminué, douleur, alanine aminotransférase augmentée et aspartate aminotransférase augmentée.
Les effets indésirables, tous grades confondus, qui ont été rapportés chez les sujets atteints de RCC et de STS ou depuis la mise sur le marché sont listés ci-dessous, par classe de systèmes d’organes, par fréquence et par grade de sévérité (classification MedDRA). La convention suivante a été utilisée pour la classification des fréquences : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Des catégories ont été définies sur la base des fréquences absolues issues des données des essais cliniques. Les données de sécurité et de tolérance post-commercialisation, issues de toutes les études cliniques réalisées avec le pazopanib et des déclarations de cas provenant de la notification spontanée, ont également été évaluées. Pour chaque classe de systèmes d’organes, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau listant les effets indésirables
Tableau 2 Effets indésirables liés au traitement rapportés dans les études RCC (n = 1 149) ou depuis la mise sur le marché
|
Classe de systèmes d’organes |
Fréquence (tous grades) |
Effets indésirables |
Tous grades n (%) |
Grade 3 n (%) |
Grade 4 n (%) |
|
Infections et Infestations |
Fréquent |
Infections (avec ou sans neutropénie) |
Fréquence indéterminée |
Fréquence indéterminée |
Fréquence indéterminée |
|
Peu fréquent |
Infection gingivale |
1 (< 1 %) |
0 |
0 |
|
|
Péritonite infectieuse |
1 (< 1 %) |
0 |
0 |
||
|
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) |
Peu fréquent |
Douleur tumorale |
1 (< 1 %) |
1 (< 1 %) |
0 |
|
Affections hématologiques et du système lymphatique |
Fréquent |
Thrombopénie |
80 (7 %) |
10 (< 1 %) |
5 (< 1 %) |
|
Neutropénie |
79 (7 %) |
20 (2 %) |
4 (< 1 %) |
||
|
Leucopénie |
63 (5 %) |
5 (< 1 %) |
0 |
||
|
Peu fréquent |
Polyglobulie |
6 (0,03 %) |
1 |
0 |
|
|
Rare |
Microangiopathie thrombotique (incluant purpura thrombotique thrombocytopénique et syndrome hémolytique et urémique) |
Fréquence indéterminée |
Fréquence indéterminée |
Fréquence indéterminée |
|
|
Affections endocriniennes |
Fréquent |
Hypothyroïdie |
83 (7 %) |
1 (< 1 %) |
0 |
|
Troubles du métabolisme et de la nutrition |
Très fréquent |
Appétit diminuée |
317 (28 %) |
14 (1 %) |
0 |
|
Fréquent |
Hypophosphatémie |
21 (2 %) |
7 (< 1 %) |
0 |
|
|
Déshydratation |
16 (1 %) |
5 (< 1 %) |
0 |
||
|
Peu fréquent |
Hypomagnésémie |
10 (< 1 %) |
0 |
0 |
|
|
Fréquence indéterminée |
Syndrome de lyse tumorale* |
Fréquence indéterminée |
Fréquence indéterminée |
Fréquence indéterminée |
|
|
Affections psychiatriques |
Fréquent |
Insomnie |
30 (3 %) |
0 |
0 |
|
Affections du système nerveux |
Très fréquent |
Dysgueusiec |
254 (22 %) |
1 (< 1 %) |
0 |
|
Céphalées |
122 (11 %) |
11 (< 1 %) |
0 |
||
|
Fréquent |
Sensations vertigineuses |
55 (5 %) |
3 (< 1 %) |
1 (< 1 %) |
|
|
Léthargie |
30 (3 %) |
3 (< 1 %) |
0 |
||
|
Paresthésie |
20 (2 %) |
2 (< 1 %) |
0 |
||
|
Neuropathie périphérique sensitive |
17 (1 %) |
0 |
0 |
||
|
Peu fréquent |
Hypoesthésie |
8 (< 1 %) |
0 |
0 |
|
|
Accident ischémique transitoire |
7 (< 1 %) |
4 (< 1 %) |
0 |
||
|
Somnolence |
3 (< 1 %) |
1 (< 1 %) |
0 |
||
|
Accident cérébrovasculaire |
2 (< 1 %) |
1 (< 1 %) |
1 (< 1 %) |
||
|
Accident ischémique cérébral |
2 (< 1 %) |
0 |
1 (< 1 %) |
||
|
Rare |
Syndrome d’encéphalopathie postérieure réversible/syndrome de leucoencéphalopathie postérieure réversible |
Fréquence indéterminée |
Fréquence indéterminée |
Fréquence indéterminée |
|
|
Affections oculaires |
Fréquent |
Vision trouble |
19 (2 %) |
1 (< 1 %) |
0 |
|
Peu fréquent |
Décollement de la rétine |
1 (< 1 %) |
1 (< 1 %) |
0 |
|
|
Déchirure rétinienne |
1 (< 1 %) |
1 (< 1 %) |
0 |
||
|
Altération de la couleur des cils |
4 (< 1 %) |
0 |
0 |
||
|
Affections cardiaques |
Peu fréquent |
Bradycardie |
6 (< 1 %) |
0 |
0 |
|
Infarctus du myocarde |
5 (< 1 %) |
1 (< 1 %) |
4 (< 1 %) |
||
|
Dysfonctionnement cardiaquef |
4 (< 1 %) |
1 (< 1 %) |
0 |
||
|
Ischémie myocardique |
3 (< 1 %) |
1 (< 1 %) |
0 |
||
|
Affections vasculaires |
Très fréquent |
Hypertension |
473 (41 %) |
115 (10 %) |
(< 1 %) |
|
Fréquent |
Bouffées de chaleur |
16 (1 %) |
0 |
0 |
|
|
Evénement thromboembolique veineuxg |
13 (1 %) |
6 (< 1 %) |
7 (< 1 %) |
||
|
Bouffées congestives |
12 (1 %) |
0 |
0 |
||
|
Peu fréquent |
Crise hypertensive |
6 (< 1 %) |
0 |
2 (< 1 %) |
|
|
Hémorragie |
1 (< 1 %) |
0 |
0 |
||
|
Rare |
Anévrismes et dissections artérielles |
Fréquence indéterminée |
Fréquence indéterminée |
Fréquence indéterminée |
|
|
Affections respiratoires, thoraciques et médiastinales |
Fréquent |
Epistaxis |
50 (4 %) |
1 (< 1 %) |
0 |
|
Dysphonie |
48 (4 %) |
0 |
0 |
||
|
Dyspnée |
42 (4 %) |
8 (< 1 %) |
1 (< 1 %) |
||
|
Hémoptysie |
15 (1 %) |
1 (< 1 %) |
0 |
||
|
Peu fréquent |
Rhinorrhée |
8 (< 1 %) |
0 |
0 |
|
|
Hémorragie pulmonaire |
2 (< 1 %) |
0 |
0 |
||
|
Pneumothorax |
1 (< 1 %) |
0 |
0 |
||
|
Rare |
Pneumopathie interstitielle diffuse/pneumopathie inflammatoire |
Fréquence indéterminée |
Fréquence indéterminée |
Fréquence indéterminée |
|
|
Affections gastro-intestinales |
Très fréquent |
Diarrhée |
614 (53 %) |
65 (6 %) |
2 (< 1 %) |
|
Nausées |
386 (34 %) |
14 (1 %) |
0 |
||
|
Vomissements |
225 (20 %) |
18 (2 %) |
1 (< 1 %) |
||
|
Douleur abdominalea |
139 (12 %) |
15 (1 %) |
0 |
||
|
Fréquent |
Stomatite |
96 (8 %) |
4 (< 1 %) |
0 |
|
|
Dyspepsie |
83 (7 %) |
2 (< 1 %) |
0 |
||
|
Flatulences |
43 (4 %) |
0 |
0 |
||
|
Distension abdominale |
36 (3 %) |
2 (< 1 %) |
0 |
||
|
Ulcération buccale |
28 (2 %) |
3 (< 1 %) |
0 |
||
|
Bouche sèche |
27 (2 %) |
0 |
0 |
||
|
Peu fréquent |
Pancréatite |
8 (< 1 %) |
4 (< 1 %) |
0 |
|
|
Hémorragie rectale |
8 (< 1 %) |
2 (< 1 %) |
0 |
||
|
Emission de selles sanglantes |
6 (< 1 %) |
0 |
0 |
||
|
Hémorragie gastro-intestinale |
4 (< 1 %) |
2 (< 1 %) |
0 |
||
|
Méléna |
4 (< 1 %) |
1 (< 1 %) |
0 |
||
|
Selles fréquentes |
3 (< 1 %) |
0 |
0 |
||
|
Hémorragie anale |
2 (< 1 %) |
0 |
0 |
||
|
Perforation du gros intestin |
2 (< 1 %) |
1 (< 1 %) |
0 |
||
|
Hémorragie buccale |
2 (< 1 %) |
0 |
0 |
||
|
Hémorragie gastro-intestinale haute |
2 (< 1 %) |
1 (< 1 %) |
0 |
||
|
Fistule entérocutanée |
1 (< 1 %) |
0 |
0 |
||
|
Hématémèse |
1 (< 1 %) |
0 |
0 |
||
|
Hémorragie hémorroïdaire |
1 (< 1 %) |
0 |
0 |
||
|
Perforation iléale |
1 (< 1 %) |
0 |
1 (< 1 %) |
||
|
Hémorragie œsophagienne |
1 (< 1 %) |
0 |
0 |
||
|
Hémorragie rétropéritonéale |
1 (< 1 %) |
0 |
0 |
||
|
Affections hépatobiliaires |
Fréquent |
Hyperbilirubinémie |
38 (3 %) |
2 (< 1 %) |
1 (< 1 %) |
|
Fonction hépatique anormale |
29 (3 %) |
13 (1 %) |
2 (< 1 %) |
||
|
Hépatotoxicité |
18 (2 %) |
11 (< 1 %) |
2 (< 1 %) |
||
|
Peu fréquent |
Ictère |
3 (< 1 %) |
1 (< 1 %) |
0 |
|
|
Lésion hépatique induite par une drogue ou un médicament |
2 (< 1 %) |
2 (< 1 %) |
0 |
||
|
Insuffisance hépatique |
1 (< 1 %) |
0 |
1 (< 1 %) |
||
|
Affections de la peau et du tissu sous-cutané |
Très fréquent |
Couleur des cheveux ou des poils modifiée |
404 (35 %) |
1 (< 1 %) |
0 |
|
Erythrodysesthésie palmo-plantaire |
206 (18 %) |
39 (3 %) |
0 |
||
|
Alopécie |
130 (11 %) |
0 |
0 |
||
|
Rash |
129 (11 %) |
7 (< 1 %) |
0 |
||
|
Fréquent |
Hypopigmentation cutanée |
52 (5 %) |
0 |
0 |
|
|
Sécheresse cutanée |
50 (4 %) |
0 |
0 |
||
|
Prurit |
29 (3 %) |
0 |
0 |
||
|
Erythème |
25 (2 %) |
0 |
0 |
||
|
Dépigmentation cutanée |
20 (2 %) |
0 |
0 |
||
|
Hyperhidrose |
17 (1 %) |
0 |
0 |
||
|
Peu fréquent |
Anomalies des ongles |
11 (< 1 %) |
0 |
0 |
|
|
Exfoliation cutanée |
10 (< 1 %) |
0 |
0 |
||
|
Réaction de photosensibilité |
7 (< 1 %) |
0 |
0 |
||
|
Rash érythémateux |
6 (< 1 %) |
0 |
0 |
||
|
Trouble cutané |
5 (< 1 %) |
0 |
0 |
||
|
Rash maculaire |
4 (< 1 %) |
0 |
0 |
||
|
Rash prurigineux |
3 (< 1 %) |
0 |
0 |
||
|
Rash vésiculaire |
3 (< 1 %) |
0 |
0 |
||
|
Prurit généralisé |
2 (< 1 %) |
1 (< 1 %) |
0 |
||
|
Rash généralisé |
2 (< 1 %) |
0 |
0 |
||
|
Rash papuleux |
2 (< 1 %) |
0 |
0 |
||
|
Erythème plantaire |
1 (< 1 %) |
0 |
0 |
||
|
Ulcère cutané |
Fréquence indéterminée |
Fréquence indéterminée |
Fréquence indéterminée |
||
|
Affections musculo-squelettiques et systémiques |
Fréquent |
Arthralgie |
48 (4 %) |
8 (< 1 %) |
0 |
|
Myalgie |
35 (3 %) |
2 (< 1 %) |
0 |
||
|
Spasmes musculaires |
25 (2 %) |
0 |
0 |
||
|
Peu fréquent |
Douleur musculo-squelettique |
9 (< 1 %) |
1 (< 1 %) |
0 |
|
|
Affections du rein et des voies urinaires |
Très fréquent |
Protéinurie |
135 (12 %) |
32 (3 %) |
0 |
|
Peu fréquent |
Hémorragie des voies urinaires |
1 (< 1 %) |
0 |
0 |
|
|
Affections des organes de reproduction et du sein |
Peu fréquent |
Ménorragie |
3 (< 1 %) |
0 |
0 |
|
Hémorragie vaginale |
3 (< 1 %) |
0 |
0 |
||
|
Métrorragie |
1 (< 1 %) |
0 |
0 |
||
|
Troubles généraux et anomalies au site d’administration |
Très fréquent |
Fatigue |
415 (36 %) |
65 (6 %) |
1 (< 1 %) |
|
Fréquent |
Inflammation muqueuse |
86 (7 %) |
5 (< 1 %) |
0 |
|
|
Asthénie |
82 (7 %) |
20 (2 %) |
1 (< 1 %) |
||
|
Œdèmeb |
72 (6 %) |
1 (< 1 %) |
0 |
||
|
Douleur thoracique |
18 (2 %) |
2 (< 1 %) |
0 |
||
|
Peu fréquent |
Frissons |
4 (< 1 %) |
0 |
0 |
|
|
Trouble muqueux |
1 (< 1 %) |
0 |
0 |
||
|
Investigations |
Très fréquent |
Alanine aminotransférase augmentée |
246 (21 %) |
84 (7 %) |
14 (1 %) |
|
Aspartate aminotransférase augmentée |
211 (18 %) |
51 (4 %) |
10 (< 1 %) |
||
|
Fréquent |
Poids diminué |
96 (8 %) |
7 (< 1 %) |
0 |
|
|
Bilirubinémie augmentée |
61 (5 %) |
6 (< 1 %) |
1 (< 1 %) |
||
|
Créatininémie augmentée |
55 (5 %) |
3 (< 1 %) |
0 |
||
|
Lipase augmentée |
51 (4 %) |
21 (2 %) |
7 (< 1 %) |
||
|
Globules blancs diminuésd |
51 (4 %) |
3 (< 1 %) |
0 |
||
|
TSH sanguine augmentée |
36 (3 %) |
0 |
0 |
||
|
Amylase augmentée |
35 (3 %) |
7 (< 1 %) |
0 |
||
|
Gamma-glutamyltransférase augmentée |
31 (3 %) |
9 (< 1 %) |
4 (< 1 %) |
||
|
Pression artérielle augmentée |
15 (1 %) |
2 (< 1 %) |
0 |
||
|
Urée sanguine augmentée |
12 (1 %) |
1 (< 1 %) |
0 |
||
|
Test hépatique anormal |
12 (1 %) |
6 (< 1 %) |
1 (< 1 %) |
||
|
Peu fréquent |
Enzyme hépatique augmentée |
11 (< 1 %) |
4 (< 1 %) |
3 (< 1 %) |
|
|
Glucose sanguin diminué |
7 (< 1 %) |
0 |
1 (< 1 %) |
||
|
Allongement de l’intervalle QT à l’électrocardiogramme |
7 (< 1 %) |
2 (< 1 %) |
0 |
||
|
Transaminases augmentées |
7 (< 1 %) |
1 (< 1 %) |
0 |
||
|
Exploration fonctionnelle de la thyroïde anormale |
3 (< 1 %) |
0 |
0 |
||
|
Pression artérielle diastolique augmentée |
2 (< 1 %) |
0 |
0 |
||
|
Pression artérielle systolique augmentée |
1 (< 1 %) |
0 |
0 |
||
|
Effet indésirable lié au traitement, rapporté depuis la mise sur le marché (déclarations de cas provenant de la notification spontanée et effets indésirables graves issus de toutes les études cliniques portant sur le pazopanib). * Effet indésirable lié au traitement rapporté uniquement depuis la mise sur le marché. La fréquence ne peut être estimée sur la base des données disponibles. Les termes suivants ont été regroupés : a Douleur abdominale, douleur abdominale haute et douleur abdominale basse b Œdème, œdème périphérique, œdème de l’œil, œdème localisé et œdème de la face c Dysgueusie, agueusie et hypogueusie d Globules blancs diminués, neutrophiles diminués et numération des leucocytes diminuée e Appétit diminué et anorexie f Dysfonctionnement cardiaque, dysfonctionnement ventriculaire gauche, insuffisance cardiaque et cardiomyopathie restrictive g Evénement thromboembolique veineux, thrombose veineuse profonde, embolie pulmonaire et thrombose |
|||||
La neutropénie, la thrombopénie et l’érythrodysesthésie palmo-plantaire ont été observées plus fréquemment chez les patients originaires de l’Asie orientale.
Tableau 3 Effets indésirables liés au traitement rapportés dans les études STS (n = 382) ou depuis la mise sur le marché
|
Classe de systèmes d’organes |
Fréquence (tous grades) |
Effets indésirables |
Tous grades n (%) |
Grade 3 n (%) |
Grade 4 n (%) |
|
Infections et infestations |
Fréquent |
Infection gingivale |
4 (1 %) |
0 |
0 |
|
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) |
Très fréquent |
Douleur tumorale |
121 (32 %) |
32 (8 %) |
0 |
|
Affections hématologiques et du système lymphatiquef |
Très fréquent |
Leucopénie |
106 (44 %) |
3 (1 %) |
0 |
|
Thrombopénie |
86 (36 %) |
7 (3 %) |
2 (< 1 %) |
||
|
Neutropénie |
79 (33 %) |
10 (4 %) |
0 |
||
|
Peu fréquent |
Microangiopathie thrombotique (incluant purpura thrombotique thrombocytopénique et syndrome hémolytique et urémique) |
1 (< 1 %) |
1 (< 1 %) |
0 |
|
|
Affections endocriniennes |
Fréquent |
Hypothyroïdie |
18 (5 %) |
0 |
0 |
|
Troubles du métabolisme et de la nutrition |
Très fréquent |
Appétit diminué |
108 (28 %) |
12 (3 %) |
0 |
|
Hypoalbuminémief |
81 (34 %) |
2 (< 1 %) |
0 |
||
|
Fréquent |
Déshydratation |
4 (1 %) |
2 (1 %) |
0 |
|
|
Peu fréquent |
Hypomagnésémie |
1 (< 1 %) |
0 |
0 |
|
|
Fréquence indéterminée |
Syndrome de lyse tumorale* |
Fréquence indéterminée |
Fréquence indéterminée |
Fréquence indéterminée |
|
|
Affections psychiatriques |
Fréquent |
Insomnie |
5 (1 %) |
1 (< 1 %) |
0 |
|
Affections du système nerveux |
Très fréquent |
Dysgueusiec |
79 (21 %) |
0 |
0 |
|
Céphalées |
54 (14 %) |
2 (< 1 %) |
0 |
||
|
Fréquent |
Neuropathie périphérique sensitive |
30 (8 %) |
1 (< 1 %) |
0 |
|
|
Sensations vertigineuses |
15 (4 %) |
0 |
0 |
||
|
Peu fréquent |
Somnolence |
3 (< 1 %) |
0 |
0 |
|
|
Paresthésie |
1 (< 1 %) |
0 |
0 |
||
|
Infarctus cérébral |
1 (< 1 %) |
0 |
1 (< 1 %) |
||
|
Affections oculaires |
Fréquent |
Vision trouble |
15 (4 %) |
0 |
0 |
|
Affections cardiaques |
Fréquent |
Dysfonctionnement cardiaqueg |
21 (5 %) |
3 (< 1 %) |
1 (< 1 %) |
|
Dysfonctionnement ventriculaire gauche |
13 (3 %) |
3 (< 1 %) |
0 |
||
|
Bradycardie |
4 (1 %) |
0 |
0 |
||
|
Peu fréquent |
Infarctus du myocarde |
1 (< 1 %) |
0 |
0 |
|
|
Affections vasculaires |
Très fréquent |
Hypertension |
152 (40 %) |
26 (7 %) |
0 |
|
Fréquent |
Evénement thromboembolique veineuxd |
13 (3 %) |
4 (1 %) |
5 (1 %) |
|
|
Bouffées de chaleur |
12 (3 %) |
0 |
0 |
||
|
Bouffées congestives |
4 (1 %) |
0 |
0 |
||
|
Peu fréquent |
Hémorragie |
2 (< 1 %) |
1 (< 1 %) |
0 |
|
|
Rare |
Anévrismes et dissections artérielles |
Fréquence indéterminée |
Fréquence indéterminée |
Fréquence indéterminée |
|
|
Affections respiratoires, thoraciques et médiastinales |
Fréquent |
Epistaxis |
22 (6 %) |
0 |
0 |
|
Dysphonie |
20 (5 %) |
0 |
0 |
||
|
Dyspnée |
14 (4 %) |
3 (< 1 %) |
0 |
||
|
Toux |
12 (3 %) |
0 |
0 |
||
|
Pneumothorax |
7 (2 %) |
2 (< 1 %) |
1 (< 1 %) |
||
|
Hoquet |
4 (1 %) |
0 |
0 |
||
|
Hémorragie pulmonaire |
4 (1 %) |
1 (< 1 %) |
0 |
||
|
Peu fréquent |
Douleur oropharyngée |
3 (< 1 %) |
0 |
0 |
|
|
Hémorragie bronchique |
2 (< 1 %) |
0 |
0 |
||
|
Rhinorrhée |
1 (< 1 %) |
0 |
0 |
||
|
Hémoptysie |
1 (< 1 %) |
0 |
0 |
||
|
Rare |
Pneumopathie interstitielle diffuse/pneumopathie inflammatoire |
Fréquence indéterminée |
Fréquence indéterminée |
Fréquence indéterminée |
|
|
Affections gastro-intestinales |
Très fréquent |
Diarrhée |
174 (46 %) |
17 (4 %) |
0 |
|
Nausées |
167 (44 %) |
8 (2 %) |
0 |
||
|
Vomissements |
96 (25 %) |
7 (2 %) |
0 |
||
|
Douleur abdominalea |
55 (14 %) |
4 (1 %) |
0 |
||
|
Stomatite |
41 (11 %) |
1 (< 1 %) |
0 |
||
|
Fréquent |
Distension abdominale |
16 (4 %) |
2 (1 %) |
0 |
|
|
Bouche sèche |
14 (4 %) |
0 |
0 |
||
|
Dyspepsie |
12 (3 %) |
0 |
0 |
||
|
Hémorragie buccale |
5 (1 %) |
0 |
0 |
||
|
Flatulences |
5 (1 %) |
0 |
0 |
||
|
Hémorragie anale |
4 (1 %) |
0 |
0 |
||
|
Peu fréquent |
Hémorragie gastro-intestinale |
2 (< 1 %) |
0 |
0 |
|
|
Hémorragie rectale |
2 (< 1 %) |
0 |
0 |
||
|
Fistule entérocutanée |
1 (< 1 %) |
1 (< 1 %) |
0 |
||
|
Hémorragie gastro-intestinale |
1 (< 1 %) |
0 |
0 |
||
|
Méléna |
2 (< 1 %) |
0 |
0 |
||
|
Hémorragie œsophagienne |
1 (< 1 %) |
0 |
1 (< 1 %) |
||
|
Péritonite |
1 (< 1 %) |
0 |
0 |
||
|
Hémorragie rétropéritonéale |
1 (< 1 %) |
0 |
0 |
||
|
Hémorragie gastro-intestinale haute |
1 (< 1 %) |
1 (< 1 %) |
0 |
||
|
Perforation iléale |
1 (< 1 %) |
0 |
1 (< 1 %) |
||
|
Affections hépatobiliaires |
Peu fréquent |
Fonction hépatique anormale |
2 (< 1 %) |
0 |
1 (< 1 %) |
|
Fréquence indéterminée |
Insuffisance hépatique* |
Fréquence indéterminée |
Fréquence indéterminée |
Fréquence indéterminée |
|
|
Affections de la peau et du tissu sous-cutané |
Très fréquent |
Couleur des cheveux ou des poils modifiée |
93 (24 %) |
0 |
0 |
|
Hypopigmentation cutanée |
80 (21 %) |
0 |
0 |
||
|
Rash avec exfoliation |
52 (14 %) |
2 (< 1 %) |
0 |
||
|
Fréquent |
Alopécie |
30 (8 %) |
0 |
0 |
|
|
Affection cutanéec |
26 (7 %) |
4 (1 %) |
0 |
||
|
Sécheresse cutanée |
21 (5 %) |
0 |
0 |
||
|
Hyperhidrose |
18 (5 %) |
0 |
0 |
||
|
Anomalies des ongles |
13 (3 %) |
0 |
0 |
||
|
Prurit |
11 (3 %) |
0 |
0 |
||
|
Erythème |
4 (1 %) |
0 |
0 |
||
|
Peu fréquent |
Ulcère cutané |
3 (< 1 %) |
1 (< 1 %) |
0 |
|
|
Rash |
1 (< 1 %) |
0 |
0 |
||
|
Rash papuleux |
1 (< 1 %) |
0 |
0 |
||
|
Réaction de photosensibilité |
1 (< 1 %) |
0 |
0 |
||
|
Erythrodysesthésie palmo-plantaire |
2 (< 1 %) |
0 |
0 |
||
|
Affections musculo-squelettiques et systémiques |
Fréquent |
Douleur musculo-squelettique |
35 (9 %) |
2 (< 1 %) |
0 |
|
Myalgie |
28 (7 %) |
2 (< 1 %) |
0 |
||
|
Spasmes musculaires |
8 (2 %) |
0 |
0 |
||
|
Peu fréquent |
Arthralgie |
2 (< 1 %) |
0 |
0 |
|
|
Affections du rein et des voies urinaires |
Peu fréquent |
Protéinurie |
2 (< 1 %) |
0 |
0 |
|
Affections des organes de reproduction et du sein |
Peu fréquent |
Hémorragie vaginale |
3 (< 1 %) |
0 |
0 |
|
Ménorragie |
1 (< 1 %) |
0 |
0 |
||
|
Troubles généraux et anomalies au site d’administration |
Très fréquent |
Fatigue |
178 (47 %) |
34 (9 %) |
1 (< 1 %) |
|
Fréquent |
Œdèmeb |
18 (5 %) |
1 (< 1 %) |
0 |
|
|
Douleur thoracique |
12 (3 %) |
4 (1 %) |
0 |
||
|
Frissons |
10 (3 %) |
0 |
0 |
||
|
Peu fréquent |
Inflammation muqueusee |
1 (< 1 %) |
0 |
0 |
|
|
Asthénie |
1 (< 1 %) |
0 |
0 |
||
|
Investigationsh |
Très fréquent |
Poids diminué |
86 (23 %) |
5 (1 %) |
0 |
|
Fréquent |
Examen des oreilles, du nez et de la gorge anormale |
29 (8 %) |
4 (1 %) |
0 |
|
|
Alanine aminotransférase augmentée |
8 (2 %) |
4 (1 %) |
2 (< 1 %) |
||
|
Cholestérol sanguin anormal |
6 (2 %) |
0 |
0 |
||
|
Aspartate aminotransférase augmentée |
5 (1 %) |
2 (< 1 %) |
2 (< 1 %) |
||
|
Gamma-glutamyltransférase augmentée |
4 (1 %) |
0 |
3 (< 1 %) |
||
|
Peu fréquent |
Bilirubinémie augmentée |
2 (< 1 %) |
0 |
0 |
|
|
Aspartate aminotransférase |
2 (< 1 %) |
0 |
2 (< 1 %) |
||
|
Alanine aminotransférase |
1 (< 1 %) |
0 |
1 (< 1 %) |
||
|
Numération plaquettaire diminuée |
1 (< 1 %) |
0 |
1 (< 1 %) |
||
|
Allongement de l’intervalle QT à l’électrocardiogramme |
2 (< 1 %) |
1 (< 1 %) |
0 |
||
|
Effet indésirable lié au traitement, rapporté depuis la mise sur le marché (déclarations de cas provenant de la notification spontanée et effets indésirables graves issus de toutes les études cliniques portant sur le pazopanib). * Effet indésirable lié au traitement rapporté uniquement depuis la mise sur le marché. La fréquence ne peut être estimée sur la base des données disponibles. Les termes suivants ont été regroupés : a Douleur abdominale, douleur abdominale haute et douleur gastro-intestinale b Œdème, œdème périphérique et œdème palpébral c La majorité de ces cas ont consisté en une érythrodysesthésie palmo-plantaire d Evénements thromboemboliques veineux incluant thrombose veineuse profonde, embolie pulmonaire et thrombose e La majorité de ces cas décrivent une mucite f Fréquence basée sur les tableaux des valeurs biologiques issus de l’étude VEG110727 (N = 240). La fréquence à laquelle ces événements indésirables ont été rapportés par les investigateurs est moins élevée que celle indiquée dans les tableaux des valeurs biologiques g Evénements liés à un dysfonctionnement cardiaque incluant dysfonctionnement ventriculaire gauche, insuffisance cardiaque et cardiomyopathie restrictive h La fréquence est basée sur les événements indésirables rapportés par les investigateurs. Les anomalies biologiques ont été rapportées en tant qu’événements indésirables par les investigateurs à une fréquence moins élevée que celle indiquée dans les tableaux des valeurs biologiques. |
|||||
La neutropénie, la thrombopénie et l’érythrodysesthésie palmo-plantaire ont été observées plus fréquemment chez les patients originaires de l’Asie orientale.
Population pédiatrique
Le profil de sécurité chez les patients pédiatriques était similaire à celui rapporté avec le pazopanib chez les adultes dans les indications approuvées sur la base des données de 44 patients pédiatriques issues de l’étude de phase I ADVL0815 et de 57 patients pédiatriques issues de l’étude de phase II PZP034X2203 (voir rubrique 5.1).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
4.9. Surdosage
Des doses de pazopanib allant jusqu’à 2 000 mg ont été évaluées dans les études cliniques. Une fatigue de grade 3 (toxicité dose-limitante) et une hypertension de grade 3 ont été l’une et l’autre observées chez 1 patient sur 3, aux doses respectives de 2 000 mg et 1 000 mg par jour.
Il n’existe pas d’antidote spécifique à un surdosage par pazopanib, le traitement devra être de soutien.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
Le pazopanib est un inhibiteur de protéine tyrosine-kinase puissant, administré par voie orale, visant plusieurs cibles : des récepteurs du facteur de croissance endothélial vasculaire (VEGFR1, VEGFR2, et VEGFR3), des récepteurs du facteur de croissance plaquettaire (PDGFRα et PDGFRβ) et le récepteur du facteur de cellule souche (c-KIT) avec des valeurs de CI50 de 10, 30, 47, 71, 84 et 74 nM respectivement. Au cours des expérimentations pré-cliniques, le pazopanib a inhibé, de façon dose-dépendante, l’auto-phosphorylation induite par la liaison avec leurs ligands, des récepteurs du VEGFR2, du c-KIT et du PDGFRβ dans les cellules. In vivo, le pazopanib a inhibé la phosphorylation du VEGFR2 induite par le VEGF dans les poumons de souris, l’angiogenèse de plusieurs modèles animaux, et la croissance de multiples greffes tumorales humaines chez la souris.
Pharmacogénomique
Dans une méta-analyse de pharmacogénétique sur les données issues de 31 études cliniques au cours desquelles le pazopanib était administré soit en monothérapie soit en association avec d’autres agents, une élévation des ALAT > 5 × LSN (NCI CTC grade 3) est survenue chez 19 % des porteurs de l’allèle HLA-B*57:01 et chez 10 % des non porteurs. Dans cet ensemble de données, 133/2 235 des patients (6 %) étaient porteurs de l’allèle HLA-B*57:01 (voir rubrique 4.4).
Etudes cliniques
Cancer du rein (RCC)
La sécurité et l’efficacité du pazopanib dans le RCC a été évaluée au cours d’une étude multicentrique, randomisée, en double-aveugle, contrôlée versus placebo. Les patients atteints d’un RCC avancé et/ou métastatique (N = 435) ont été randomisés pour recevoir 800 mg de pazopanib une fois par jour ou un placebo. L’objectif principal de l’étude était d’évaluer et de comparer la survie sans progression (SSP) dans les deux bras de traitement et le critère d’efficacité secondaire majeur était la survie globale (SG). Les autres objectifs étaient d’évaluer le taux de réponse globale et la durée de la réponse.
Au total, sur les 435 patients de l’étude, 233 patients étaient naïfs de traitement et 202 étaient des patients de deuxième ligne ayant préalablement reçu un traitement à base d’interleukine-2 ou d’interféron α. L’indice de performance (ECOG) était similaire entre les groupes pazopanib et placebo (ECOG 0 : 42 % vs 41 %, ECOG 1 : 58 % vs 59 %). La majorité des patients avait des facteurs pronostiques de Motzer/MSKCC (Memorial Sloan Kettering Cancer Centre) soit favorables (39 %), soit intermédiaires (54 %). Tous les patients avaient un cancer du rein de type histologique à cellules claires ou à prédominance de cellules claires. Près de la moitié des patients avaient 3 organes ou plus atteints et la plupart des patients avaient, à l’inclusion, des métastases pulmonaires (74 %) et/ou ganglionnaires (54 %).
Le nombre de patients naïfs de traitement et prétraités par des cytokines était équivalent dans chaque bras de traitement (53 % et 47 % dans le bras pazopanib, 54 % et 46 % dans le bras placebo). Dans le sous-groupe des patients prétraités par des cytokines, la majorité (75 %) avait reçu un traitement à base d’interféron.
Un pourcentage similaire de patients dans chaque bras de traitement, a subi préalablement une néphrectomie (89 % et 88 % dans les bras pazopanib et placebo respectivement) et/ou a reçu préalablement une radiothérapie (22 % et 15 % dans les bras pazopanib et placebo, respectivement).
L’analyse principale du critère d’évaluation primaire (SSP) était basée sur l’évaluation de la maladie par une revue radiologique indépendante dans la totalité de la population de l’étude (patients naïfs de traitement et prétraités par des cytokines).
Tableau 4 Résultats d’efficacité globale dans le RCC par une évaluation indépendante (VEG105192)
|
Critères d’évaluation/Population de l’étude |
Pazopanib |
Placebo |
RR (IC à 95 %) |
Valeur de P (unilatérale) |
|
SSP Population en ITT globale* Médiane (mois) |
N = 290 9,2 |
N = 145 4,2 |
0,46 (0,34 ; 0,62) |
< 0,0000001 |
|
Taux de réponse % (IC à 95 %) |
N = 290 30 (25,1 ; 35,6) |
N = 145 3 (0,5 ; 6,4) |
– |
< 0,001 |
|
RR = rapport de risque ; ITT = intention de traiter ; SSP = survie sans progression. * populations de patients naïfs de traitement et prétraités par des cytokines |
||||
Figure 1 Courbe de Kaplan-Meier de la survie sans progression selon une évaluation indépendante dans la population globale (populations de patients naïfs de traitement et prétraités par des cytokines) (VEG105192)
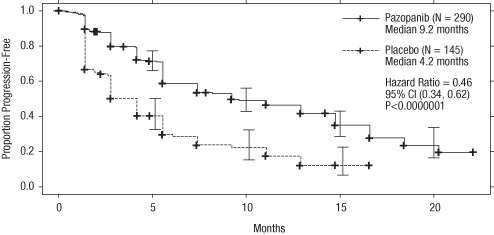
Axe des abscisses : Mois ; axe des ordonnées : Pourcentage sans progression ; Pazopanib (N = 290) ; Médiane = 9,2 mois ; Placebo (N = 145) ; Médiane = 4,2 mois ; Rapport de risque = 0,46 ; IC à 95 % (0,34 ; 0,62) ; P < 0,0000001
Figure 2 Courbe de Kaplan-Meier de la survie sans progression selon une évaluation indépendante dans la population de patients naïfs de traitement (VEG105192)
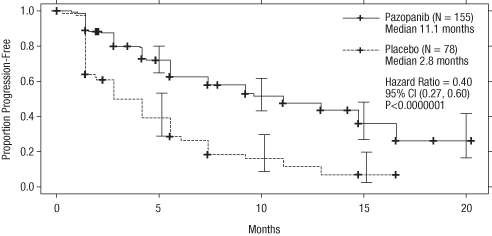
Axe des abscisses : Mois ; axe des ordonnées : Pourcentage sans progression ; Pazopanib (N = 155) ; Médiane = 11,1 mois ; Placebo (N = 78) ; Médiane = 2,8 mois ; Rapport de risque = 0,40 ; IC à 95 % (0,27 ; 0,60) ; P < 0,0000001
Figure 3 Courbe de Kaplan-Meier de la survie sans progression selon une évaluation indépendante dans la population de patients prétraités par des cytokines (VEG105192)
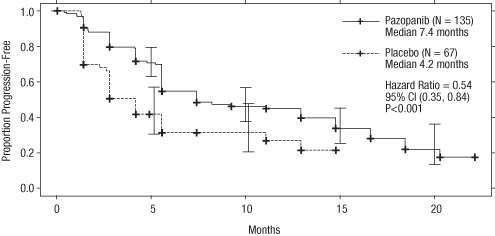
Axe des abscisses : Mois ; axe des ordonnées : Pourcentage sans progression ; Pazopanib (N = 135) ; Médiane = 7,4 mois ; Placebo (N = 67) ; Médiane = 4,2 mois ; Rapport de risque = 0,54 ; IC à 95 % (0,35 ; 0,84) ; P < 0,001
Pour les patients ayant répondu au traitement, le temps de réponse (médiane) était de 11,9 semaines et la durée de réponse (médiane) était de 58,7 semaines selon l’évaluation indépendante (VEG105192).
La médiane de survie globale lors de l’analyse finale de survie prévue par le protocole était de 22,9 mois et 20,5 mois (RR = 0,91 [IC 95 % : 0,71 ; 1,16 ; p = 0,224]) pour les patients randomisés respectivement dans les bras pazopanib et placebo. Les résultats de SG peuvent potentiellement être biaisés étant donné que 54 % des patients du bras placebo ont également reçu du pazopanib dans le cadre de l’extension de cette étude suite à la progression de leur maladie. Soixante-six pour cent des patients sous placebo ont reçu un traitement post-étude par rapport à 30 % des patients sous pazopanib.
Aucune différence statistique n’a été observée entre les groupes de traitement concernant la qualité de vie globale évaluée par les échelles EORTC QLQ-C30 et EuroQoL EQ-5D.
Dans une étude de phase II portant sur 225 patients présentant un cancer du rein à cellules claires récidivant localement ou métastatique, le taux de réponse objective était de 35 % et la durée de réponse (médiane) était de 68 semaines, selon une évaluation indépendante. La SSP (médiane) était de 11,9 mois.
La sécurité, l’efficacité et la qualité de vie du pazopanib versus sunitinib étaient évaluées au cours d’une étude de non-infériorité de phase III, randomisée, en ouvert et en groupes parallèles (VEG108844).
Dans l’étude VEG108844, les patients (N = 1 110) atteints d’un RCC localement avancé et/ou métastatique et n’ayant pas reçu au préalable de traitement par voie systémique, étaient randomisés afin de recevoir soit du pazopanib à la dose de 800 mg une fois par jour en continu soit du sunitinib 50 mg une fois par jour selon des cycles de traitement de 6 semaines avec 4 semaines de traitement suivies de 2 semaines sans traitement.
L’objectif principal de cette étude était d’évaluer et de comparer la SSP entre les patients traités par pazopanib et ceux traités par sunitinib. Les caractéristiques démographiques étaient similaires entre les bras de traitement. Les caractéristiques de la maladie déterminées lors du diagnostic initial et à la sélection étaient équilibrées entre les bras de traitement, la majorité des patients ayant un cancer à cellules claires et à un stade IV.
L’étude VEG108844 a atteint son objectif principal sur la SSP et a démontré la non-infériorité du pazopanib par rapport au sunitinib, car la borne supérieure de l’IC de 95 % pour le rapport de risque était inférieure à la borne de non infériorité de 1,25 spécifiée dans le protocole. Les résultats d’efficacité globale sont résumés dans le tableau 5.
Tableau 5 Résultats d’efficacité globale (VEG108844)
|
Critères d’évaluation |
Pazopanib N = 557 |
Sunitinib N = 553 |
HR (IC 95 %) |
|
PFS |
|
|
|
|
Globale |
|
|
|
|
Médiane (mois) (IC 95 %) |
8,4 (8,3 ; 10,9) |
9,5 (8,3 ; 11,0) |
1,047 (0,898 ; 1,220) |
|
Survie globale Médiane (mois) (IC 95 %) |
28,3 (26,0 ; 35,5) |
29,1 (25,4 ; 33,1) |
0,915a (0,786 ; 1,065) |
|
RR = rapport de risque ; SSP = survie sans progression ; a valeur de P = 0,245 (bilatérale) |
|||
Figure 4 Courbe de Kaplan-Meier pour la survie sans progression dans la population globale selon une évaluation indépendante (VEG108844)
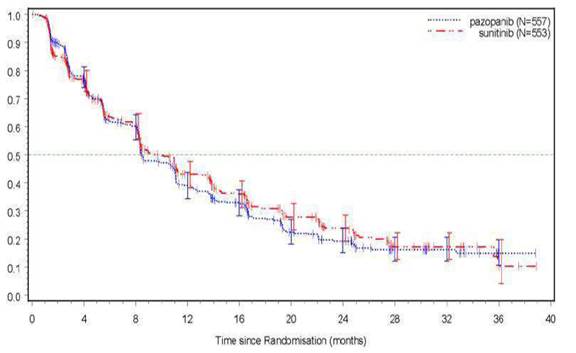
Axe des abscisses : Temps depuis la randomisation (mois) ; pazopanib (N = 557) ; sunitinib (N = 553)
Des analyses de SSP en sous-groupes ont été réalisées sur 20 facteurs pronostiques et démographiques. Les intervalles de confiance à 95 % pour l’ensemble des sous-groupes incluent un rapport de risque de 1. L’estimation du rapport de risque dépassait 1,25 dans les trois plus petits de ces 20 sous-groupes, c’est-à-dire chez les sujets n’ayant pas eu de néphrectomie préalable (n = 186, RR = 1,403, IC à 95 % [0,955 ; 2,061]), ayant des LDH à l’inclusion > 1,5 × LSN (n = 68, RR = 1,72, IC à 95 % [0,943 ; 3,139]), et chez ceux du groupe pronostic MSKCC défavorable (n = 119, RR = 1,472, IC à 95 % [0,937 ; 2,313]).
Sarcome des tissus mous (STS)
L’efficacité et la sécurité du pazopanib dans le STS ont été évaluées dans une étude pivot de phase III multicentrique, randomisée, en double aveugle, contrôlée versus placebo (VEG110727). Au total, 369 patients ayant un STS avancé étaient randomisés pour recevoir du pazopanib à la dose de 800 mg une fois par jour ou un placebo. Il est important de noter que seuls les patients présentant des sous-types histologiques spécifiques de STS ont été autorisés à participer à l’étude ; par conséquent, la sécurité et l’efficacité du pazopanib ne peuvent être considérées comme établies que pour ces sous-groupes et le traitement par pazopanib doit se limiter à ces seuls sous-types de STS.
Les types de tumeur suivants étaient éligibles :
Les tumeurs fibroblastiques (fibrosarcome de l’adulte, myxofibrosarcome, fibrosarcome épithélioïde sclérosant, tumeurs malignes fibreuses solitaires), les tumeurs dites fibrohistiocytaires (histiocytome fibreux malin pléomorphe [MFH], MFH à cellules géantes, MFH inflammatoire), les léiomyosarcomes, les tumeurs glomiques malignes, les tumeurs des muscles squelettiques (rhabodmyosarcome pléomorphique et alvéolaire), les tumeurs vasculaires (hémangioendothéliome épithélioïde, angiosarcome), les tumeurs de différentiation incertaine (synoviales, épithélioïdes, alvéolaires des parties molles, à cellules claires, à petites cellules rondes desmoplastiques, rhabdoïdes extra-rénales, mésenchymome malin, PEComes, sarcome intimal), les tumeurs malignes de la gaine des nerfs périphériques, les sarcomes des tissus mous indifférenciés sans autre indication (SAI) et les autres types de sarcome (non listés comme étant inéligibles).
Les types de tumeurs suivants étaient non éligibles :
Les liposarcomes (tous les sous-types), tous les rhabdomyosarcomes qui n’étaient ni alvéolaires ni pléomorphiques, les chondrosarcomes, les ostéosarcomes, les tumeurs d’Ewing/tumeurs neuroectodermiques primitives (PNET), les GIST, les dermatofibrosarcomes protuberans, les sarcomes myofibroblastiques inflammatoires, le mésothéliome malin et les tumeurs mésodermiques mixtes de l’utérus.
Il est important de souligner que les patients ayant un liposarcome ont été exclus de l’étude pivot de phase III car dans l’étude préliminaire de phase II (VEG20002), l’activité observée avec le pazopanib dans le liposarcome (SSP à la Semaine 12) n’a pas atteint le taux pré-requis pour poursuivre les évaluations cliniques.
Les autres critères principaux d’éligibilité de l’étude VEG110727 étaient un STS de haut grade de malignité ou de grade intermédiaire prouvé histologiquement et une progression de la maladie dans les 6 mois suivant un traitement pour maladie métastatique ou une rechute dans les 12 mois suivant un traitement (néo) adjuvant.
Quatre-vingt-dix-huit pour cent (98 %) des sujets avaient préalablement reçu de la doxorubicine, 70 % de l’ifosfamide et 65 % des sujets avaient reçu au moins trois agents de chimiothérapie avant d’entrer dans l’étude.
Les patients étaient stratifiés selon l’indice de performance de l’OMS (IP OMS) (0 ou 1) à l’inclusion ainsi que selon le nombre de lignes de traitements systémiques antérieurs reçus pour la maladie avancée (0 ou 1 vs 2+). Dans chaque groupe de traitement, un pourcentage légèrement supérieur de sujets avait reçu au moins 2 lignes de traitements systémiques antérieurs pour leur maladie avancée (respectivement, 58 % et 55 % dans les bras placebo et pazopanib) par rapport aux sujets ayant reçu 0 ou 1 ligne de traitements systémiques antérieurs (respectivement, 42 % et 45 % dans les bras placebo et pazopanib). La durée médiane de suivi des sujets (définie de la date de randomisation à la date du dernier contact ou décès) était similaire dans les deux bras de traitement (9,36 mois pour le bras placebo [intervalle de 0,69 à 23,0 mois] et 10,04 mois pour le bras pazopanib [intervalle de 0,2 à 24,3 mois]).
L’objectif principal de cette étude était la survie sans progression (SSP évaluée par une revue radiologique indépendante) ; les critères d’évaluation secondaires incluaient la survie globale (SG), le taux de réponse globale et la durée de la réponse.
Tableau 6 Résultats d’efficacité globale dans le STS selon une évaluation indépendante (VEG110727)
|
Critères d’évaluation/population de l’étude |
Pazopanib |
Placebo |
RR (IC à 95 %) |
Valeur de P (bilatérale) |
|
SSP |
|
|
|
|
|
Population en ITT globale |
N = 246 |
N = 123 |
|
|
|
Médiane (semaines) |
20,0 |
7,0 |
0,35 (0,26 ; 0,48) |
< 0,001 |
|
Léiomyosarcomes |
N = 109 |
N = 49 |
|
|
|
Médiane (semaines) |
20,1 |
8,1 |
0,37 (0,23 ; 0,60) |
< 0,001 |
|
Sous-groupes de synovialosarcomes |
N = 25 |
N = 13 |
|
|
|
Médiane (semaines) |
17,9 |
4,1 |
0,43 (0,19 ; 0,98) |
0,005 |
|
Sous-groupes d’« Autres STS » |
N = 112 |
N = 61 |
|
|
|
Médiane (semaines) |
20,1 |
4,3 |
0,39 (0,25 ; 0,60) |
< 0,001 |
|
SG |
|
|
|
|
|
Population en ITT globale |
N = 246 |
N = 123 |
|
|
|
Médiane (mois) |
12,6 |
10,7 |
0,87 (0,67 ; 1,12) |
0,256 |
|
Léiomyosarcomes* |
N = 109 |
N = 49 |
|
|
|
Médiane (mois) |
16,7 |
14,1 |
0,84 (0,56 ; 1,26) |
0,363 |
|
Sous-groupes de synovialosarcomes* |
N = 25 |
N = 13 |
|
|
|
Médiane (mois) |
8,7 |
21,6 |
1,62 (0,79 ; 3,33) |
0,115 |
|
Sous-groupes d’« Autres STS »* |
N = 112 |
N = 61 |
0,84 (0,59 ; 1,21) |
|
|
Médiane (mois) |
10,3 |
9,5 |
|
0,325 |
|
Taux de réponse (RC + RP) % (IC à 95 %) |
4 (2,3 ; 7,9)
|
0 (0,0 ; 3,0) |
|
|
|
Durée de la réponse Médiane (semaines) (IC à 95 %) |
38,9 (16,7 ; 40,0) |
|
|
|
|
RR = rapport de risque ; ITT = intention de traiter ; SSP = survie sans progression ; RC = réponse complète ; RP = réponse partielle ; SG = survie globale * La survie globale pour les sous-groupes histologiques respectifs de STS (léiomyosarcome, synovialosarcome et « autres » STS) doit être interprétée avec prudence en raison du faible nombre de sujets et des intervalles de confiance larges |
||||
Une amélioration similaire de la SSP basée sur les évaluations des investigateurs a été observée dans les bras pazopanib et placebo (dans la population en ITT globale, RR : 0,39 ; IC à 95 % [0,30 ; 0,52], p < 0,001).
Figure 5 Courbe de Kaplan-Meier de la survie sans progression dans le STS évaluée d’après une revue indépendante pour la population globale (VEG110727)
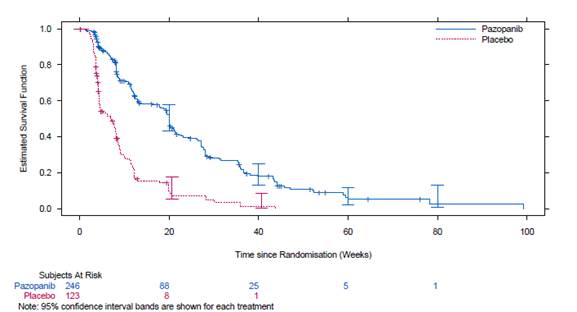
Axe des abscisses : Temps depuis la randomisation (semaines) ; axe des ordonnées : Fonction de survie estimée ; Pazopanib ; Placebo ; Sujets à risque ; Pazopanib 246 88 25 5 1 ; Placebo 123 8 1 ; Remarque : les bandes d’intervalle de confiance à 95 % sont indiquées pour chaque traitement
L’analyse finale de la SG effectuée à 76 % (280/369) des événements n’a montré aucune différence significative entre les deux bras de traitement (RR 0,87, IC à 95 % [0,67 ; 1,12] p = 0,256).
Population pédiatrique
Une étude de phase I (ADVL0815) portant sur le pazopanib a été menée chez 44 patients pédiatriques atteints de diverses tumeurs solides récurrentes ou réfractaires. L’objectif principal était d’étudier la dose maximale tolérée (DMT), le profil de sécurité et les propriétés pharmacocinétiques du pazopanib chez l’enfant. La durée médiane d’exposition dans cette étude était de 3 mois (1– 23 mois).
Une étude de phase II (PZP034X2203) portant sur le pazopanib a été menée chez 57 patients pédiatriques atteints de tumeurs solides réfractaires incluant rhabdomyosarcome (N = 12), sarcome des tissus mous hors rhabdomyosarcome (N = 11), sarcome d’Ewing/pPNET (N = 10), ostéosarcome (N = 10), neuroblastome (N = 8) et hépatoblastome (N = 6). L’étude en monothérapie, en ouvert, non contrôlée, était destinée à déterminer l’activité thérapeutique du pazopanib chez les enfants et adolescents âgés de 1 à <18 ans. Le pazopanib était administré quotidiennement sous forme de comprimé à la dose de 450 mg/m2/dose ou sous forme de solution buvable à 225 mg/m2/dose. La dose quotidienne maximale autorisée était de 800 mg pour le comprimé et de 400 mg pour la solution buvable. La durée médiane d’exposition était de 1,8 mois (1 jour– 29 mois).
Les résultats de cette étude n’ont pas révélé d’activité antitumorale significative dans cette population pédiatrique. Le pazopanib n’est donc pas recommandé pour le traitement de ces tumeurs dans la population pédiatrique (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec le médicament de référence contenant du pazopanib dans tous les sous-groupes de la population pédiatrique dans le cancer du rein et du carcinome pelvien rénal (à l’exception de néphroblastome, néphroblastomatose, sarcome à cellules claires, néphrome mésoblastique, carcinome médullaire rénal et tumeur rhabdoïde du rein) (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
5.2. Propriétés pharmacocinétiques
Absorption
Suite à une administration par voie orale d’une dose unique de pazopanib de 800 mg à des patients ayant des tumeurs solides, la concentration plasmatique maximale (Cmax), obtenue après une médiane de 3,5 heures (intervalle de 1,0 à 11,9 heures) était approximativement de 19 ± 13 μg/mL et l’ASC0-∞ était approximativement de 650 ± 500 μg.h/mL. Des administrations journalières entraînent une augmentation de l’ASC0-T d’un facteur 1,23 à 4.
Il n’y a pas eu d’augmentation importante de l’ASC ou de la Cmax pour des doses de pazopanib supérieures à 800 mg.
L’exposition systémique au pazopanib est augmentée lors d’une administration avec de la nourriture. L’administration de pazopanib au cours d’un repas riche ou pauvre en matières grasses a entraîné une augmentation de l’ASC et de la Cmax d’un facteur 2 environ. Par conséquent, le pazopanib doit être administré au moins deux heures après ou une heure avant un repas (voir rubrique 4.2).
L’administration de comprimé écrasé dosé à 400 mg de pazopanib a augmenté l’ASC(0-72) de 46 %, la Cmax d’un facteur 2 environ et a diminué le tmax d’approximativement 2 heures par rapport à l’administration d’un comprimé entier. Ces résultats montrent que la biodisponibilité et la vitesse d’absorption orale du pazopanib sont augmentées après l’administration d’un comprimé écrasé par rapport à l’administration d’un comprimé entier (voir rubrique 4.2).
Distribution
La liaison du pazopanib aux protéines plasmatiques humaines in vivo était de plus de 99 % quelle que soit la concentration comprise dans un intervalle de 10 à 100 µg/mL. Les études in vitro suggèrent que le pazopanib est un substrat pour la glycoprotéine P (P-gp) et la BCRP.
Biotransformation
Les résultats d’études in vitro ont démontré que le métabolisme du pazopanib était médié principalement par le CYP3A4, avec des contributions mineures du CYP1A2 et du CYP2C8. Les quatre principaux métabolites du pazopanib représentent seulement 6 % de l’exposition plasmatique. L’un de ces métabolites inhibe la prolifération des cellules endothéliales de la veine ombilicale humaine stimulées par le VEGF avec une puissance similaire à celle du pazopanib.
Les autres sont 10 à 20 fois moins actifs. Par conséquent, l’activité du pazopanib est principalement dépendante de l’exposition au pazopanib parent.
Elimination
Le pazopanib est éliminé lentement avec une demi-vie moyenne de 30,9 heures après administration de la dose recommandée de 800 mg. L’élimination se fait principalement par les fèces avec une élimination rénale comptant pour < 4 % de la dose administrée.
Populations particulières
Insuffisance rénale
Les résultats indiquent que moins de 4 % de la dose de pazopanib administrée par voie orale est excrétée dans l’urine sous forme de pazopanib et de métabolites. Les résultats issus d’un modèle de pharmacocinétique de population (données provenant de sujets ayant, à l’inclusion, des valeurs de clairance de la créatinine comprises entre 30,8 mL/min et 150 mL/min) ont indiqué qu’il était peu probable qu’une insuffisance rénale ait un effet cliniquement significatif sur la pharmacocinétique du pazopanib. Aucune adaptation posologique n’est requise chez les patients ayant une clairance de la créatinine supérieure à 30 mL/min. Etant donné qu’aucune donnée n’est disponible avec le pazopanib chez les patients ayant une clairance de la créatinine inférieure à 30 mL/min, il est conseillé d’être prudent avec cette population de patients (voir rubrique 4.2).
Insuffisance hépatique
Légère
Chez les patients présentant de légères anomalies des paramètres hépatiques (définies, soit par un taux normal de bilirubine associé à une augmentation du taux d’alanine aminotransférase [ALAT], quel qu’en soit le degré, soit par une augmentation du taux de bilirubine jusqu’à 1,5 × LSN, indépendamment du taux d’ALAT), les valeurs médianes de Cmax et d’ASC(0-24) à l’équilibre après administration de 800 mg de pazopanib une fois par jour sont similaires aux valeurs médianes observées chez les patients présentant une fonction hépatique normale (voir tableau 7). Huit cents milligrammes de pazopanib une fois par jour constitue la posologie recommandée chez les patients présentant de légères anomalies du bilan hépatique sérique (voir rubrique 4.2).
Modérée
La dose maximale tolérée (DMT) de pazopanib chez les patients présentant une insuffisance hépatique modérée (définie par une augmentation de la bilirubine > 1,5 × 3 × LSN indépendamment du taux d’ALAT) était de 200 mg une fois par jour. Les valeurs médianes, à l’état d’équilibre, de la Cmax et de l’ASC(0-24) après administration de 200 mg de pazopanib une fois par jour chez des patients présentant une insuffisance hépatique modérée étaient d’environ 44 % et 39 % des valeurs médianes correspondantes obtenues après administration de 800 mg de pazopanib une fois par jour chez des patients présentant une fonction hépatique normale (voir tableau 7).
Au vu des données de sécurité et de tolérance, la posologie du pazopanib doit être réduite à 200 mg une fois par jour chez les sujets présentant une insuffisance hépatique modérée (voir rubrique 4.2).
Sévère
Les valeurs médianes, à l’état d’équilibre, de la Cmax et de l’ASC(0-24) après administration de 200 mg de pazopanib une fois par jour chez des patients présentant une insuffisance hépatique sévère étaient d’environ 18 % et 15 % des valeurs médianes correspondantes obtenues après administration de 800 mg de pazopanib une fois par jour chez des patients présentant une fonction hépatique normale. Sur la base d’une exposition diminuée et d’une réserve hépatique limitée, le pazopanib n’est pas recommandé chez les patients présentant une insuffisance hépatique sévère (définie par une bilirubine totale > 3 × LSN indépendamment du taux d’ALAT) (voir rubrique 4.2).
Tableau 7 Médiane des paramètres pharmacocinétiques à l’état d’équilibre mesurés chez des sujets insuffisants hépatiques
|
Groupe |
Dose à l’étude |
Cmax(µg/mL) |
ASC(0-24) (µg × h/mL) |
Dose recommandée |
|
Fonction hépatique normale |
800 mg 1x/j |
52,0 (17,1– 85,7) |
888,2 (345,5– 1 482) |
800 mg 1x/j |
|
IH légère |
800 mg 1x/j |
33,5 (11,3– 104,2) |
774,2 (214,7– 2 034,4) |
800 mg 1x/j |
|
IH modérée |
200 mg 1x/j |
22,2 (4,2– 32,9) |
256,8 (65,7– 487,7) |
200 mg 1x/j |
|
IH sévère |
200 mg 1x/j |
9,4 (2,4– 24,3) |
130,6 (46,9– 473,2) |
Non recommandé |
|
1x/j : une fois par jour |
||||
Population pédiatrique
Lors de l’administration de pazopanib 225 mg/m2 (sous forme de solution buvable) à des patients pédiatriques, les paramètres pharmacocinétiques (Cmax, Tmax et ASC) étaient similaires à ceux précédemment rapportés chez les patients adultes traités par 800 mg de pazopanib. Les résultats n’ont indiqué aucune différence notable dans la clairance du pazopanib, calculée en fonction de la surface corporelle, entre les enfants et les adultes.
5.3. Données de sécurité préclinique
Le profil de sécurité préclinique du pazopanib a été évalué chez des souris, des rats, des lapins et des singes. Dans les études à doses répétées chez les rongeurs, il a été identifié que les effets sur plusieurs types de tissus (os, dent, lit de l’ongle, organes reproducteurs, tissus hématologiques, rein et pancréas) semblent liés à la pharmacologie de l’inhibition du VEGFR et/ou à la perturbation des signaux du VEGF, avec des effets plus importants survenant à des taux d’exposition plasmatique en dessous des niveaux observés en clinique. Les autres effets observés incluent une perte de poids, des diarrhées et/ou des morbidités soit secondaires à des effets gastro-intestinaux locaux dus à une forte exposition du médicament au niveau de la muqueuse locale (singes), soit à des effets pharmacologiques (rongeurs). Sur la base de l’ASC, des lésions hépatiques prolifératives (foyers éosinophiliques et adénomes) ont été mises en évidence chez les souris femelles, lors d’expositions 2,5 fois plus importantes que l’exposition chez l’être humain.
Dans les études de toxicité chez le jeune animal, au cours desquelles les rats en période de pré-sevrage recevaient du pazopanib du 9e au 14e jour après la naissance, des décès ainsi qu’une croissance/maturation anormale des organes au niveau du rein, du poumon, du foie, et du cœur, ont été observés, avec des doses d’environ 0,1 fois les doses cliniques (doses basées sur l’ASC et utilisées chez l’être humain adulte). Chez les rats en période de post-sevrage qui recevaient du pazopanib du 21e au 62e jour après la naissance, des anomalies toxicologiques similaires à celles observées chez les rats adultes et à des doses comparables ont été observées. Chez les patients pédiatriques humains, il existe un risque accru d’effets sur les os et les dents dans la population pédiatrique par rapport aux adultes, étant donné que ces effets, y compris l’inhibition de la croissance (membres plus courts), os fragilisés, et remodelage des dents, ont été constatés chez le jeune rat à des doses ≥ 10 mg/kg/jour (équivalentes à environ 0,1– 0,2 fois les doses cliniques, basées sur l’ASC et utilisées chez l’être humain adulte) (voir rubrique 4.4).
Effets sur la reproduction, la fertilité et le développement du fœtus
Il a été démontré que le pazopanib était embryotoxique et tératogène lorsqu’il était administré chez des rats et des lapins à des expositions plus de 300 fois inférieures à l’imprégnation chez l’être humain (sur la base de l’ASC). Les effets incluent une réduction de la fertilité chez la femelle, une augmentation des risques de perte de l’embryon avant et après implantation, des résorptions précoces, une létalité embryonnaire, une diminution du poids du fœtus et des malformations cardiovasculaires. Une diminution de la formation du corps jaune, une augmentation des kystes et une atrophie des ovaires ont également été observées chez les rongeurs. Dans une étude sur la fertilité chez le rat mâle, aucun effet n’a été mis en évidence sur l’accouplement ou la fertilité ; cependant, une diminution du poids des testicules et de l’épididyme a été observée avec des réductions des taux de production de sperme, de la motilité des spermatozoïdes et des concentrations de sperme dans les testicules et l’épididyme observés à des imprégnations plasmatiques 0,3 fois supérieures à l’exposition chez l’être humain (sur la base de l’ASC).
Génotoxicité
Aucune altération génétique n’a été mise en évidence avec le pazopanib lors des essais de génotoxicité (test d’Ames, test d’aberration chromosomique sur lymphocytes périphériques humains et test du micronucleus in vivo chez le rat). Un intermédiaire de fabrication synthétique du pazopanib, qui est également présent dans la substance active finale en faibles quantités, n’était pas mutagénique lors du test d’Ames mais a révélé un potentiel génotoxique au cours du test de lymphome de souris et du test du micronucleus chez la souris in vivo.
Carcinogénicité
Au cours des études de carcinogénicité menées sur deux ans avec le pazopanib, il a été constaté une augmentation du nombre d’adénomes hépatiques chez les souris et d’adénocarcinomes duodénaux chez les rats. Sur la base de la pathogenèse spécifique aux rongeurs et du mécanisme de ces événements, ils ne sont pas considérés comme représentant une augmentation du risque carcinogène chez les patients sous pazopanib.
6. DONNEES PHARMACEUTIQUES
6.1. Liste des excipients
Noyau du comprimé :
Cellulose microcristalline (E460), carboxyméthylamidon sodique (type A), povidone K30 (E1201), stéarate de magnésium (E470b).
Pelliculage :
Hypromellose (E464), dioxyde de titane (E171), macrogol 400 (E1521), polysorbate 80 (E433).
6.2. Incompatibilités
6.3. Durée de conservation
48 mois.
6.4. Précautions particulières de conservation
6.5. Nature et contenu de l'emballage extérieur
30, 60 comprimés pelliculés en flacons (PEHD) blancs munis d’un bouchon sécurité-enfant en polypropylène blanc.
30, 60, conditionnement multiple de 60 (2 boîtes de 30), 90, conditionnement multiple de 90 (3 boîtes de 30) comprimés pelliculés sous plaquettes (Aluminium-PVC/PE/PVDC) transparentes.
30 × 1, 60 × 1, conditionnement multiple de 60 × 1 (2 boîtes de 30 × 1), 90 × 1, conditionnement multiple de 90 × 1 (3 boîtes de 30 × 1) comprimés pelliculés sous plaquettes prédécoupées unitaires (Aluminium-PVC/PE/PVDC) transparentes.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 513 7 9 : 30 comprimés sous plaquettes (Aluminium/PVC/PE/PVDC).
· 34009 302 513 8 6 : 60 comprimés sous plaquettes (Aluminium/PVC/PE/PVDC).
· 34009 302 513 9 3 : Conditionnement multiple de 60 comprimés (2 boîtes de 30) comprimés sous plaquettes (Aluminium/PVC/PE/PVDC).
· 34009 302 514 0 9 : 30 x1 comprimés sous plaquettes prédécoupées unitaires (Aluminium/PVC/PE/PVDC).
· 34009 302 514 1 6 : 60 x1 comprimés sous plaquettes prédécoupées unitaires (Aluminium/PVC/PE/PVDC).
· 34009 302 514 2 3 : Conditionnement multiple de 60 (2 boîtes de 30 x1) comprimés sous plaquettes (Aluminium/PVC/PE/PVDC).
· 34009 302 514 3 0 : 30 comprimés en flacon (PEHD).
· 34009 302 514 4 7 : 60 comprimés en flacon (PEHD).
· 34009 550 890 6 3 : 90 comprimés sous plaquettes (Aluminium/PVC/PE/PVDC).
· 34009 550 890 7 0 : Conditionnement multiple de 90 comprimés (3 boîtes de 30) comprimés sous plaquettes (Aluminium/PVC/PE/PVDC).
· 34009 550 890 8 7 : 90 x1 comprimés sous plaquettes prédécoupées unitaires (Aluminium/PVC/PE/PVDC).
· 34009 550 891 0 0 : Conditionnement multiple de 90 (3 boîtes de 30 x1) comprimés sous plaquettes (Aluminium/PVC/PE/PVDC).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation:{JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
11. DOSIMETRIE
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament soumis à prescription hospitalière.
Prescription réservée aux spécialistes en oncologie ou en hématologie ou aux médecins compétents en cancérologie.
Médicament nécessitant une surveillance particulière pendant le traitement.